

 English
English文献解读|Cell Rep Med(14.3):启动子高甲基化导致的基因沉默塑造透明细胞肾细胞癌的肿瘤微环境和免疫治疗耐药性
✦ +
+
论文ID
原名:Silencing of genes by promoter hypermethylation shapes tumor microenvironment and resistance to immunotherapy in clear-cell renal cell carcinomas
译名:启动子高甲基化导致的基因沉默塑造透明细胞肾细胞癌的肿瘤微环境和免疫治疗耐药性
期刊:Cell Reports Medicine
影响因子:14.3
发表时间:2023.11.21
DOI号:10.1016/j.xcrm.2023.101287
背 景
透明细胞肾细胞癌 (ccRCC) 是主要的肾癌,导致许多癌症相关死亡。目前的指南建议一线治疗采用双重免疫检查点抑制 (ICI) 或血管内皮生长因子 (VEGF) 受体酪氨酸激酶抑制剂 (TKI) 和抗 PD-1 ICI 的组合。IMmotion150 和 JAVELIN Renal 101 等实验的转录组学特征显示,Ipi/Nivo 和 nivolumab 单一疗法的预测价值有限,这使得 ccRCC 免疫疗法的生物标志物鉴定具有挑战性。
实验设计
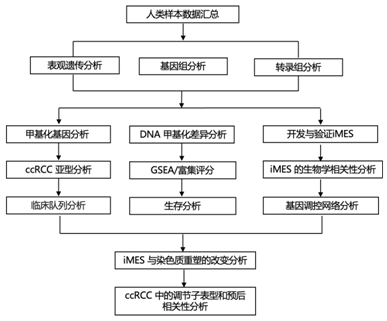
结 果
01

表观遗传沉默与SETD2突变、9p 缺失和肿瘤侵袭性相关
使用 Illumina 450K 阵列,研究者团队研究了来自癌症基因组图谱 (TCGA) 的 301 个 ccRCC 肿瘤和 160 个正常肾脏样本(图 1)。整合分析确定了 493 个基因通过启动子 CpG 岛 (CGI) 处的癌症特异性 DNA 高甲基化而发生表观遗传沉默。
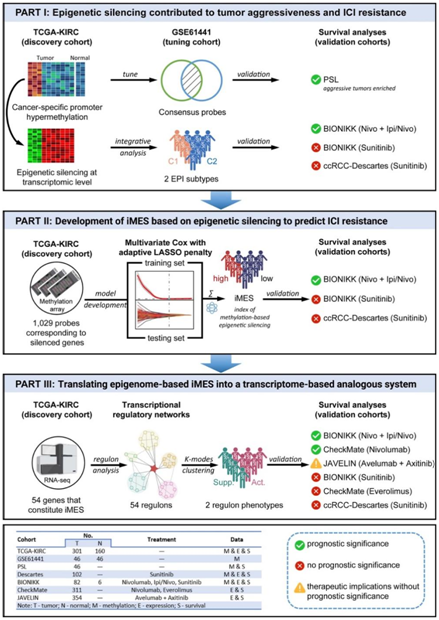
图1. 研究流程图。
通过对 101 个频繁甲基化基因 (>20%) 进行无监督聚类,他们确定了两种 ccRCC 亚型:EPI-C1和 EPI-C2(图 2 A)。与 EPI-C2 相比,EPI-C1具有更高比例的增强子去甲基化表型 (TED+)表型,这两种亚型与患者的临床结果密切相关(图 2 B)。值得注意的是,通过单变量分析,101个沉默基因中有75个与预后相关,并且所有的基因都与死亡风险增加相关,这表明表观遗传沉默在ccRCC中具有广泛的预后相关性。
相比于EPI-C2,EPI-C1与晚期肿瘤分级(G1 + G2;G3 + G4)和分期(ⅰ+ⅱ;ⅲ+ⅳ)密切相关(图2C-D)。他们发现在29例肉瘤样和横纹肌样分化的ccRCC(sccRCC/rccRCC)中,有13例属于EPI-C1,表明EPI-C1在sccRCC中显著富集于EPI-C2。
鉴于9p缺失与肾癌侵袭性之间的相关性,他们检测了TCGA-KIRC队列,发现EPI-C1的9p缺失比EPI-C2更常见(图2A)。此外,与EPI-C2相比,SET Domain Containing 2 (SETD2)是唯一在EPI-C1中频繁富集突变的基因。
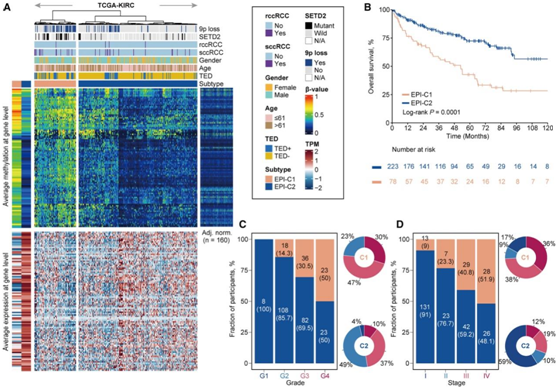
图2. DNA甲基化导致的表观遗传沉默与ccRCC肿瘤侵袭性的关系。
(A) TCGA队列中甲基化和基因表达谱的热图,包括邻近正常组织和临床特征的注释。(B) Kaplan-Meier曲线描绘了表观遗传亚型的OS率。(C)柱状图和饼状图显示了两种表观遗传亚型与肿瘤分级之间的关联。(D)与(C)相同,不同的是肿瘤分期。
02
表观遗传沉默会导致EZH2过度表达、PRC2高甲基化和BAP1缺失
然后他们检测了两种表观遗传亚型之间的 DNA 甲基化差异。在 TCGA-KIRC 队列中,他们在 EPI-C1 中发现了 2363 个高甲基化探针,而在 EPI-C2 中仅发现了 14 个。这些主要位于启动子 CGI 中,并富含 Polycomb 抑制复合物 2 (PRC2) 靶标。值得注意的是,与 EPI-C2 相比,EPI-C1 中 Polycomb 靶点的超甲基化与 zeste 2 Polycomb 抑制复合物 2 亚基 (EZH2) 增强子的过度表达相一致(图3A)。EZH2是PRC2的关键组成部分,其过度表达可导致PRC2活性增加,从而导致靶基因(包括PRC2本身)的高甲基化。
有趣的是,EZH2的过度表达与BAP1 的缺失有关。他们确定了BAP1突变肿瘤中染色质可及性降低的区域,并验证了BAP1缺失驱动的染色质抑制特征 (BAP1-LCR)。该特征与EZH2表达呈负相关,并且在 EPI-C1 中减弱(图3 B),表明BAP1丢失可能不仅仅包括突变,还可能导致在 ccRCC 中观察到表观遗传沉默。
基因集富集分析(GSEA)显示,TCGA-KIRC队列中的EPI-C1与炎症反应、上皮-间质转化(EMT)和IL-6/JAK/STAT3 Hallmark通路的表达增加有关(图3C)。EPI-C1中肉瘤样蛋白的富集与EMT在肉瘤样和横纹肌样变化中的作用一致。IL-6/JAK/STAT3通路的激活表明EPI-C1具有更高的免疫逃逸可能性,并具有较高的肿瘤免疫功能障碍和排斥(TIDE)评分(图3D-E)。
他们的综合分析确定了 341 个沉默基因,其中 101 个频繁发生甲基化。在这些基因中,有 21 个与 TCGA-KIRC 队列中确定的基因重叠(图3F)。值得注意的是,他们观察到这些基因的细胞遗传学区域 19q13.43 显著富集,其中包括锌指蛋白,例如ZNF135、ZNF154、ZNF471、ZNF835和ZIK1。通过监督聚类,他们在接受 ICI 治疗的患者中确定了两种表观遗传亚型(EPI-C1;EPI-C2)(图 3 G)。EPI-C1 患者的总生存期 (OS) 显著较差(图 3 H)。与 EPI-C1 相比,EPI-C2 的无进展生存期 (PFS)和第二次治疗时间 (TST)几乎是 EPI-C1 的两倍。与EPI-C2相比,他们还观察到EPI-C1亚组中9p缺失的边际富集,特别是在9p21.3的病灶区域(图3G)。同样,他们发现BAP1-LCR特征与EZH2表达呈负相关, EPI-C1的特征评分低于EPI-C2(图3G)。这些结果表明,表观遗传沉默可能在ccRCC的侵袭性和对ICI的抗性形成中发挥作用。
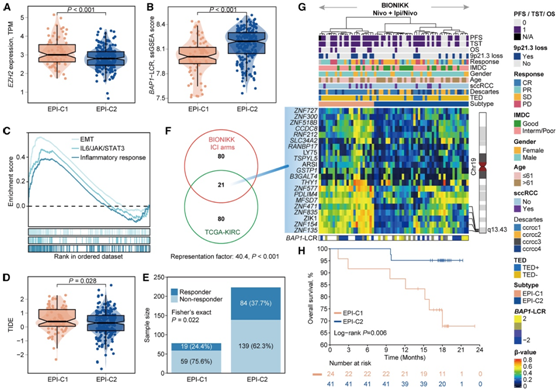
图3. DNA甲基化导致的表观遗传沉默及其在ccRCC ICI治疗原发耐药中的作用。
(A) TCGA 队列表观遗传亚型之间 EZH2 表达的小提琴图。(B) TCGA 队列表观遗传亚型之间BAP1 -LCR 水平的小提琴图。(C) TCGA 队列 EPI-C1 激活途径的 GSEA。(D) TCGA 队列表观遗传亚型之间 TIDE 评分的小提琴图。(E) TCGA 队列的两种表观遗传亚型中 TIDE 预测的 ICI 应答者分数的堆积条形图。(F) TCGA-KIRC 和 BIONIKK 的 ICI 臂之间表观遗传沉默基因交叉的维恩图。(G) BIONIKK ICI 臂中两种表观遗传亚型中表观遗传沉默基因的 DNA 甲基化景观。(H) Kaplan-Meier 曲线描绘了 BIONIKK ICI 组中表观遗传亚型的 OS 率。
03
与 ICI 耐药性相关的 iMES 的开发和验证
使用具有自适应最小绝对收缩和选择算子(adaLASSO)的多变量Cox回归模型,他们基于10倍交叉验证确定了当偏似然偏差达到最小值时的最优λ值(图4A)。选择了58个adaLASSO系数为非零的探针,用于建立基于甲基化的表观遗传沉默指数(iMES)(图4B)。他们通过这些探针的线性组合计算每个样本的iMES,并按其系数加权。根据TCGA-KIRC队列训练集的iMES将患者分为iMES-高组和iMES-低组。在这一组中,较高的iMES与较差的OS显著相关。然后,他们将iMES应用于整个TCGA-KIRC队列,由此显示出与OS的显著关联(图4C)。时间依赖的受试者工作特征(ROC)分析表明,iMES具有良好的判别能力,1年、3年、5年和10年的时间依赖曲线下面积(AUC)值分别为0.86、0.86、0.87和0.91(图4D)。
为了评估iMES作为接受ICI治疗患者预后指标的潜力,BIONIKK队列中计算了每个样本的iMES。iMES能够将患者分为iMES-高和iMES-低组,PFS、TST和OS的预后有显著差异(图4E-G)。与iMES-低组相比,iMES-高组的ICI无应答者显著增加。无应答者的iMES显著高于应答者。时间依赖性ROC分析显示,iMES对短期和长期OS的预测均具有良好的区分能力(平均AUC为0.91),但对TST的预测能力仅限于短期(3个月内的平均AUC为0.83)(图4H)。
然后他们研究了 iMES 是否可以作为局部和转移性 ccRCC 的独立预后因素(图 4 I)。在调整 TCGA-KIRC 队列中的其他主要临床预后特征后,iMES 仍然是一个重要的独立预后因素。这些发现表明 iMES 有可能增强国际转移性肾细胞癌数据库联盟的预测风险评分,特别是对于接受免疫治疗的患者。
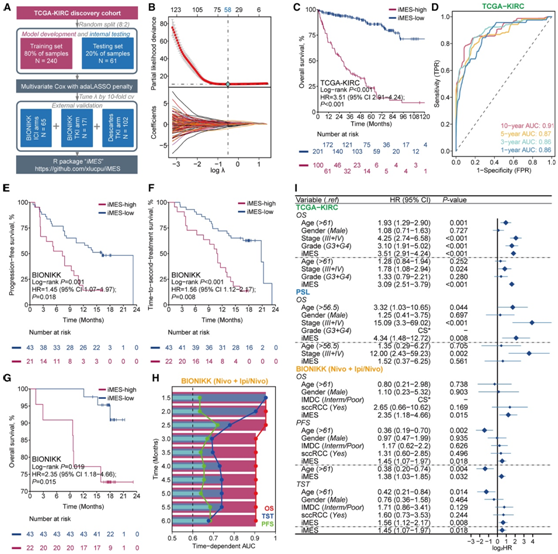
图4. iMES 的开发和验证。
(A) 流程图。(B) adaLASSO 模型中 λ 的选择。(C) Kaplan-Meier 生存曲线描绘了 TCGA 队列中两个 iMES 组的 OS 率。(D) TCGA 队列中 iMES 的时间依赖性 ROC 曲线。(E) BIONIKK ICI 组中通过 iMES 分层的患者 PFS 的 Kaplan-Meier 曲线。(F-G) 与 (E) 相同,但适用于 TST。(H) BIONIKK ICI 组治疗后 6 个月内 iMES 的时间依赖性 ROC 曲线。(I) 森林图显示单变量(虚线上方)和多变量(虚线下方)分析中的风险比。
04
iMES 与高免疫浸润和上皮损耗相关
为了破译 iMES 的生物学基础,他们对 TCGA-KIRC 队列和 BIONIKK ICI 组中具有高和低 iMES 的样本进行了 GSEA。高 iMES 样本显著富集与炎症反应、干扰素 γ 反应、EMT 和 IL-6/JAK/STAT3 信号传导相关的通路。在癌细胞系百科全书 (CCLE) 数据集中也发现了类似的趋势(图 5 A)。鉴于干扰素-γ通路在高iMES患者中的激活,他们检测了ccRCC样本中免疫检查点基因的表达,并研究了两个队列的特异性免疫细胞浸润状态和TME的功能方向(图5B-C)。用 TR4(由上皮细胞、内皮细胞、成纤维细胞和大量免疫细胞群组成的特征矩阵)进一步反卷积(图 5D)揭示了 iMES 和免疫细胞之间的正相关性,而与两个队列中的上皮细胞呈负相关。同样,在两个队列中,与低 iMES 的患者相比,高 iMES 的患者具有更高比例的免疫细胞和更低的内皮细胞(图 5D)。

图5. iMES的生物学相关性。
(A) GSEA 分析显示具有高 iMES 的患者/细胞系中激活的 Hallmark 通路。(B) TCGA 队列的 TME 景观,样本根据 iMES 按升序排序。(C) 与 (B) 相同,但适用于 BIONIKK 的 ICI 臂。(D) iMES 和细胞分数之间的关联。
05
内皮耗竭与 VEGF 通路甲基化一致
在高iMES的ccRCC患者中,内皮细胞特征的减少凸显了探索VEGF通路在血管生成和肿瘤进展中的重要性。他们发现4个VEGF相关基因- FLT4、PRKCB、FLT1和KDR-频繁甲基化并且在它们的甲基化模式中同时出现(图6A)。排列试验表明,iMES与这些基因的甲基化呈正相关。FLT4基因在DNA甲基化和表达之间具有最高的负相关(图6B)。在BIONIK的ICI组中,相同的VEGF基因经常发生共甲基化(图6A),包括FLT4(32.3%)、PRKCB(72.3%)、FLT1(41.5%)和KDR(38.5%)。其中,FLT4、PRKCB和KDR与iMES呈正相关。他们还注意到DNA甲基化与FLT4和PRKCB的表达呈负相关(图6B)。
他们在 TCGA-KIRC 队列中确定了 6 个与 iMES 表现出强正相关的调节因子,包括EZH2 、KAT2A、KDM5D、KMT5A、PRDM14和SIRT7(图6C)。值得注意的是,KMT5A和EZH2与 BIONIKK ICI 组中的 iMES 呈正相关(图 6C),这与EZH2在 BIONIKK ICI 组中的过度表达一致。 此外,他们发现在 TCGA-KIRC 队列中,iMES 与SETD2的调节子活性呈负相关,表明高 iMES 患者的SETD2丢失。BIONIKK 队列和 TCGA-KIRC 队列中的SETD2丢失特征与 iMES 呈正相关(图6C)。
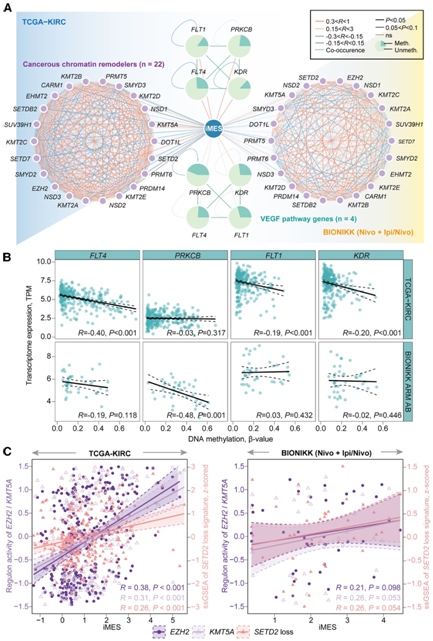
图6. iMES 与 VEGF 和染色质重塑的关联。
(A) 网络说明 iMES(蓝点)和VEGF通路基因的甲基化状态(绿色饼图)和癌性染色质重塑因子的调节子活性(紫色点)之间的关联,以及染色质重塑之间的内部相关性、相互排斥状态VEGF通路基因(连接VEGF基因的绿线),以及它们在基因水平 DNA 甲基化和基因表达之间的自相关性(饼图周围的定向弯曲线)。(B) 四个VEGF通路基因的基因水平甲基化和表达之间的相关性。(C) iMES 与两个染色质重塑因子(EZH2和KMT5A)的调节子活性之间的相关性,以及 iMES 与STED2丢失特征的单样本 GSEA 评分之间的相关性。
06
将基于表观基因组的 iMES 转化为基于转录组的类似系统
在 TCGA-KIRC 队列中,他们发现了两种不同的表型,表现出不同的调节子活动模式(图 7 A)。与调节子激活表型相比,调节子抑制表型表现出显著较高的 iMES,并且在 iMES 高组患者中比例过高(图 7 B)。根据类似系统,显示下调调节子模式的表型将表示沉默状态,类似于 iMES-高组。相反,调节子模式上调的表型表明处于活跃的非沉默状态,反映了 iMES-低组。生存分析表明 OS 具有显著的预后相关性,调节子抑制表型的中位生存期为 63.7 个月,而调节子激活表型的中位生存期尚未达到(图 7 C)。
为了进一步探讨类似系统对接受 ICI 治疗的 ccRCC 患者的预后能力,他们将分析范围扩展到接受 Ipi/Nivo 或 nivolumab 单药治疗的 BIONIKK 和 CheckMate 队列。在这些队列中,两种调节子表型成功复制,与调节子激活表型的患者相比,调节子抑制表型的患者表现出明显较差的预后。
在 BIONIKK ICI 组中(图 7D),具有调节子抑制表型的患者表现出显著更短的 TST和 PFS(图7E-F)。
在 CheckMate ICI 组中(图 7G),调节子抑制表型在 OS 方面表现出明显较差的结果(图 7H),并且这种表型由免疫激活/抑制群体高度渗透,但内皮细胞发生耗尽,这与与调节子激活表型相比血管生成/基质和血管生成亚型的富集一致(图 7 G)。反卷积分析显示,与调节子激活表型相比,调节子抑制表型中免疫细胞的比例较高,但内皮细胞的比例较低(图 7 G)。
在对JAVELIN队列的探索中,患者接受了ICI和TKI (avelumab axitinib)的联合治疗,这与BIONIKK和CheckMate队列中的治疗不同,这两种调节子表型显示出与先前观察到的相似的模式(图7I)。调控因子抑制表型的特征是免疫浸润增加,内皮细胞减少,EZH2和KMT5A活性增强,SETD2和BAP1缺乏。同样,他们注意到在调控激活表型中血管生成/基质和血管生成聚类的显著富集。然而,就表型而言,PFS没有统计学意义(图7J)。这一结果说明这些患者可能对ICI + TKI治疗有反应。
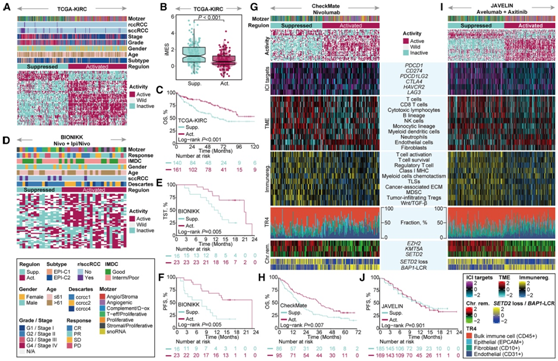
图7. ccRCC 中的调节子表型和预后相关性。
(A)根据 TCGA 队列中的调节子活性状态,对与模型选择的探针相对应的基因进行k均值聚类。(B) 两种调节子亚型之间的 iMES 小提琴图。(C) Kaplan-Meier 生存曲线分析。(D)使用 BIONIKK 的 ICI 臂中的调节子活性状态进行K均值聚类。(E) Kaplan-Meier 曲线区分 BIONIKK ICI 臂中调节子亚型的 TST。(F) 与 (E) 相同,但针对 PFS。(G) k均值聚类,使用调控子活性状态(上)、细胞分数反卷积的TME图景(中)、CheckMate ICI分支中EZH2、KMT5A和SETD2的调控子活性分布以及SETD2损失特征的单样本GSEA评分(下)。(H) Kaplan-Meier生存曲线描绘了CheckMate ICI臂中调控子亚型的OS率。(I) JAVELIN的ICI/TKI臂中调控子表型的表征。(J) Kaplan-Meier生存曲线描绘了JAVELIN的ICI/TKI组中调节亚型的PFS率。
+ + + + + + + + + + +
结 论
本项研究整合表观遗传(DNA 甲基化)和转录组数据来识别 ccRCC 亚型,其特征是癌症特异性启动子高甲基化和 Polycomb 靶标的表观遗传沉默。他们开发并验证了基于甲基化的表观遗传沉默 (iMES) 指数,该指数可预测 BIONIKK 试验中对免疫检查点抑制 (ICI) 的主要耐药性。高 iMES 与VEGF通路沉默、内皮细胞耗竭、免疫激活/抑制、EZH2激活、BAP1 / SETD2缺陷和 ICI 抵抗相关。与低甲基化药物或酪氨酸激酶抑制剂联合治疗可能有益于高 iMES 患者。有趣的是,低 iMES 的肿瘤表现出内皮细胞增加和 ICI 反应改善,这表明血管生成在 ICI 治疗中的重要性。他们还开发了一种基于转录组的类似系统,以扩展 iMES 的适用性。本项研究强调了表观遗传改变和肿瘤微环境在决定免疫治疗反应中的相互作用。
+ + + + +



