

 English
English文献解读|Cell(64.5):小细胞肺癌的蛋白质组学特征确定了生物学见解和亚型特异性治疗策略
✦ +
+
论文ID
原名:Proteogenomic characterization of small cell lung cancer identifies biological insights and subtype-specific therapeutic strategies
译名:小细胞肺癌的蛋白质组学特征确定了生物学见解和亚型特异性治疗策略
期刊:Cell
影响因子:64.5
发表时间:2024.01.04
DOI号:10.1016/j.cell.2023.12.004
背 景
肺癌是全世界癌症死亡的主要原因。小细胞肺癌 (SCLC) 占所有肺癌的 15%,是最恶性和最致命的亚型。SCLC 是一种侵袭性神经内分泌 (NE) 癌,具有快速增殖、强转移倾向和显著的治疗耐药性,这导致其预后极差。SCLC 在临床病理学、生物学和治疗选择方面与非小细胞肺癌 (NSCLC) 形成鲜明对比。在过去的几十年里,患者的生存率并没有显著改善,SCLC 仍然处于精准医疗领域之外。
实验设计
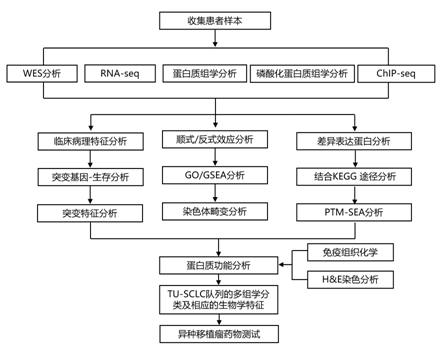
结 果
01

中国人群 SCLC 蛋白质基因组分析概述
为了描述中国 SCLC 的蛋白质组状况,研究者团队根据标准化方案收集了 112 个未经治疗的原发性 SCLC 肿瘤和来自手术切除的配对正常邻近组织 (NAT)(同济大学 [TU]-SCLC 队列),并使用全外显子组测序 (WES)、RNA测序(RNA-seq) 和整体蛋白质组学和磷酸化蛋白质组学进行分析(图1A)。总共鉴定出 28471 个非同义体细胞突变和 27080 个体细胞拷贝数改变 (SCNA),以及鉴定出11209 个蛋白质和62881 个可靠定位的磷酸位点。TU-SCLC 队列中最常见的突变基因是TP53 (72%) 和RB1 (56%)(图 1 B)。染色质修饰酶(KMT2C、KMT2D、CREBBP)、原钙粘蛋白基因(FAT1、FAT4)、转录调节因子(ZFHX3、NCOR2、CRTC1)和NOTCH家族基因(NOTCH1/2/3)的突变频率均大于10%。通过对拷贝数变异的综合分析,发现只有9例(8%)肿瘤在基因组水平上没有TP53和RB1的改变(图 1 B),表明TP53和RB1失活在SCLC肿瘤发生中的重要作用。尽管TP53和RB1失活在SCLC中占主导地位,但TU-SCLC队列中TP53的突变率相对低于主要来自西方人群的队列,但与来自日本队列相当(图 1 C)。TU-SCLC队列RB1突变率低于George等队列,但与其他两个队列无显著差异。此外,他们观察到TU-SCLC队列中ZFHX3的高突变频率(19%)(图 1 C)。
TU-SCLC 的中位肿瘤突变负荷 (TMB) 为每百万碱基对 5.45 个非同义突变。根据该中位截断值确定TMB-high和TMB-low,TMB-high组的TP53、KMT2C、FAT4和GNAS突变率显著高于TMB-low组。TMB-high的患者往往有更好的总生存期 (OS)(图 1 D)。
他们根据突变三核苷酸序列基序的频率确定了四个突变特征 (Sig1-4)。烟草诱变剂暴露与 Sig1 和 Sig2 最为匹配,有缺陷的DNA错配修复 (dMMR) 与 Sig3 相关,而 Sig4 则具有APOBEC胞苷脱氨酶的特征。根据突变特征贡献,将93例(83%)患者归类为吸烟为主导因素(smoking-dominant),17例(15%)为错配修复缺陷主导型(dMMR-dominant),只有2例(2%)为APOBEC主导型(APOBEC-dominant)(图 1 E)。
重要的是,dMMR-dominant的患者表现出更差的OS(图 1 F),而smoking-dominant肿瘤的TMB和新抗原负荷明显更高(图1G)。与smoking-dominant肿瘤相比,dMMR-dominant肿瘤中TP53突变较少,FAT1和错配修复(MMR)途径基因(MSH2、MSH6、PMS3)突变较多(图1H)。这些结果强调了烟草致癌物与SCLC肿瘤发生的关系,并证明了DNA修复缺陷在SCLC发展中的重要作用。
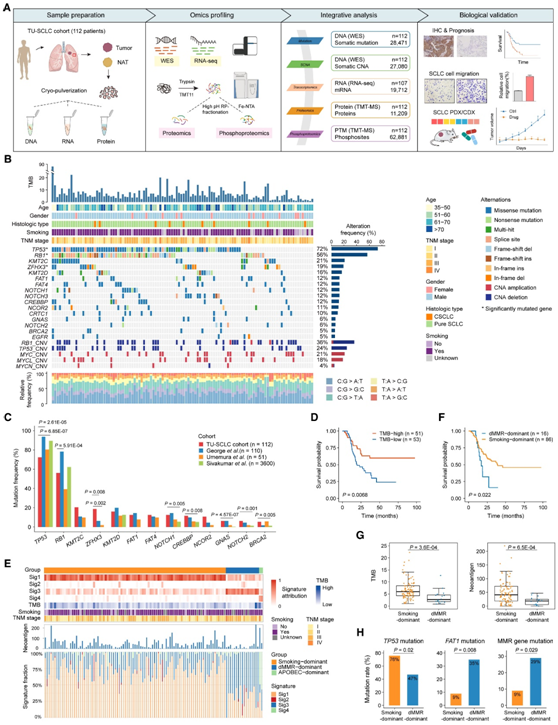
图1. TU-SCLC队列的基因组图谱。
(A) SCLC 蛋白质组分析的实验工作流程。(B)所有 112 名 SCLC 患者的基因谱和相关临床病理特征。(C) TU-SCLC 队列与其他已发表的 SCLC 队列之间突变频率的比较。(D) 基于 TMB(对数秩检验)的总体生存 Kaplan-Meier 曲线。(E) 基于突变特征比例的 SCLC 肿瘤聚类。(F) smoking-dominant和 dMMR-dominant的患者总生存率的 Kaplan-Meier 曲线。(G) smoking-dominant和 dMMR-dominant肿瘤之间 TMB 和预测新抗原的比较。(H) smoking-dominant和 dMMR-dominant肿瘤之间TP53、FAT1和 MMR 相关基因(MSH2、MSH6和PMS2 )突变率的比较。
02
通过综合分析研究基因组畸变的影响
SCNA分析发现染色体1p、1q、3q、5p、8q、14p、14q、18p和18q有扩增,而染色体3p、4p、4q、5q、13q、15q、16q、17p和17q有缺失。他们在顺式和反式效应中检测了SCNA对mRNA、蛋白质和磷蛋白丰度的影响(图2A)。在mRNA、蛋白和磷蛋白上分别观察到5363、2177和386个显著正顺式相关(图2B)。在所有三个组学重叠的261个显著顺式效应中,223个蛋白在肿瘤和NATs之间表现出不同的丰度,这些蛋白在mRNA加工、DNA修复和有丝分裂细胞周期过程的调控中富集(图2C)。基于593种癌症相关基因(CAG)的参考列表,他们发现了类似的衰减趋势(图2B)。在染色体5q上观察到有反式关联的CNA区域(图2A)。多数反式效应与5q呈负相关,且从mRNA到蛋白表达呈明显衰减。这些5q抗相关蛋白在包括有丝分裂细胞周期相变在内的多个生物过程中发生聚合。5q缺失与参与DNA复制、DNA修复和细胞周期进展的基因的高表达有关。
他们专注于鉴定含有前10个蛋白水平反式事件的cag。其中,NKX2-1(又称甲状腺转录因子1 [TTF-1])是肺癌的标志物,GNAS、AURKA、SRC、TOP1等多个CNA基因是SCLC的原癌基因或药物靶点。值得注意的是,肿瘤抑制因子RB1主要在蛋白质水平上显示拷贝数缺失反式效应,而在mRNA水平上几乎没有。在691个显著反式影响蛋白中,524个与RB1缺失正相关,富集于粒细胞活化、干扰素应答、肌动蛋白丝组织和细胞黏附等途径,而其他167个负相关蛋白主要参与DNA构象改变、mRNA加工和RNA剪接等途径(图2E-F)。磷酸蛋白水平上的进一步反式效应证实了这些途径受到RB1缺失的影响。
接下来,他们研究了体细胞突变对其同源基因产物的影响,并确定了三个具有显著顺式效应的基因(TP53、FAT1和GNAS)。与NAT相比,FAT1野生型(WT)肿瘤中的FAT1蛋白表达显著下调,尽管mRNA丰度升高(图2G)。基于蛋白质组学的基因集富集分析(GSEA)将FAT1突变与上皮间充质转化(EMT)和黏着斑途径的上调联系起来(图2H)。参与EMT、间充质分化、整合素、细胞外基质(ECM)和黏着斑途径的蛋白在fat1突变肿瘤中上调(图2I)。此外,磷酸化蛋白质组学数据显示,在fat1突变的肿瘤中,24个VIM磷酸化位点增加(图2J),这些升高的磷酸化位点主要参与细胞骨架重组和细胞运动过程。
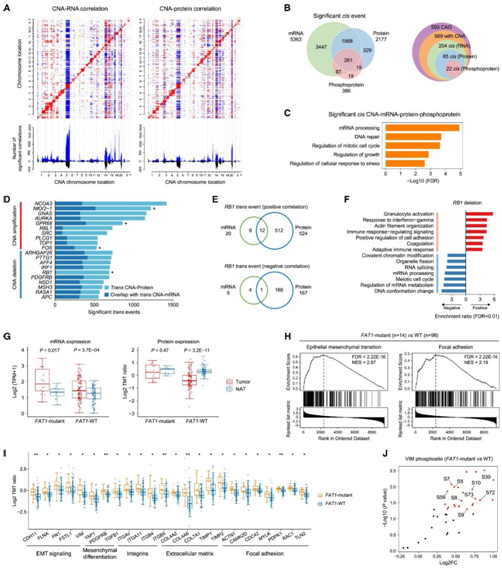
图2. SCLC 的体细胞改变和突变谱。
(A) CNA 与 mRNA(左)和蛋白质(右)丰度的相关性。(B) 维恩图描绘了SCNA(CAG,癌症相关基因)的顺式级联效应。(C)GO分析。(D) 条形图显示mRNA 和蛋白质水平的反式相关事件总数。(E) 维恩图,描绘了与RB1缺失具有正(上)或负(下)反式CNA-mRNA 和 CNA-蛋白质相关性的mRNA/蛋白质。(F)与RB1缺失具有正或负反式CNA 蛋白相关性的蛋白的 KEGG 通路富集。(G)分别比较FAT1突变体和FAT1 -WT组的肿瘤和NAT之间的mRNA和蛋白质丰度。(H) FAT1-突变体与FAT1 -WT 比较中上皮间质转化 (EMT) 和黏着斑途径的 GSEA 图。(I) FAT1突变体和FAT1 WT 肿瘤之间注释通路的蛋白质丰度比较。(J) FAT1-突变体和FAT1 -WT肿瘤之间VIM亚磷酸盐丰度的比较。
03
肿瘤-NAT 比较揭示 SCLC 相关蛋白质组事件
主成分分析 (PCA) 显示肿瘤和 NAT 在 mRNA、蛋白质和磷酸化水平上存在明显差异,并揭示肿瘤相对于 NAT 具有更大的异质性。他们重点关注没有缺失值的蛋白质和磷酸位点的丰度,并对肿瘤和 NAT 之间进行了差异分析(图 3A-F)。差异表达蛋白的富集分析显示,肿瘤中DNA复制、剪接体、细胞周期和DNA修复等途径上调,补体和凝血级联、ecm受体相互作用、黏着斑、胆固醇代谢和花生四烯酸代谢途径下调(图3B)。他们重点研究了25个蛋白,它们在90%以上的肿瘤- nat对中表达量升高2倍以上,他们将其定义为sclc相关蛋白(图3C)。这些蛋白包括微管动力学相关蛋白STMN1/2、DNA损伤修复蛋白TMA7和PCNA,以及转录调节因子(HMGB3、PHF6、DDX5和SUPT16H)与患者生存率呈负相关。实验表明,STMN1和TMA7的敲低显著降低了H345细胞系的细胞增殖,提示它们在SCLC中的致癌作用。在纯SCLC以及合并SCLC (CSCLC)的肿瘤- nat比较中,大多数蛋白质的变化相似(图3D)。然而,22种主要参与NE相关过程的蛋白在纯SCLC肿瘤中特异性增加(图3E)。此外,他们观察到1667个磷酸化位点在肿瘤中与NAT相比上调2倍以上,其中1357个(81%)磷酸化位点丰度的变化大于相应蛋白丰度的变化(图3G)。
接下来,他们根据其底物的磷酸化水平和相应的激酶激活位点推断出激酶活性。磷酸基特异性特征富集分析(PTM-SEA)鉴定出SCLC中19个活性升高的激酶(图3H),其中5个激酶的激活位点表达升高,包括CHEK1、ATR、ATM、CDK2和GSK3A(图3I)。此外,这些激酶在肿瘤- nat比较中磷酸化丰度的变化大于相应蛋白丰度的变化。已知CHEK1在基因毒性应激反应中对G2/M检查点至关重要。他们发现CHEK1蛋白及其S317磷酸化位点水平在肿瘤中升高,同时参与细胞周期阻滞和DNA损伤反应(DDR)的底物如CDC25C S216、TP53BP1 S1678和TP53 S15的表达升高(图3J)。
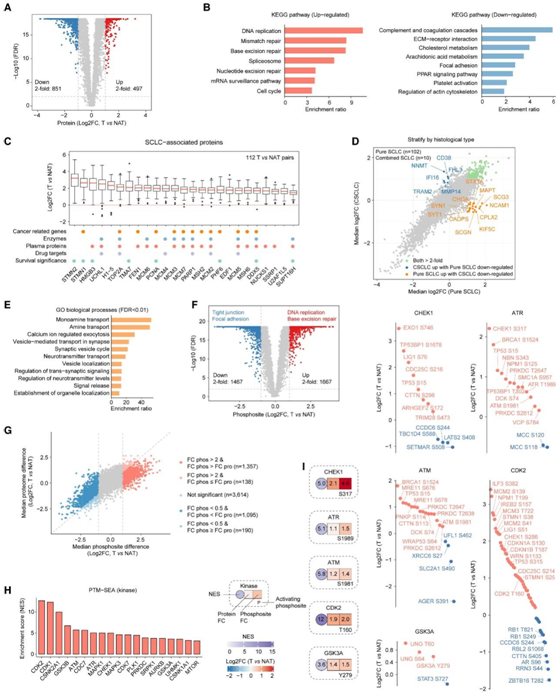
图3. 与肿瘤发生和预后相关的蛋白质基因组改变。
(A) 火山图显示肿瘤和配对 NAT 之间差异表达的蛋白质。(B) 蛋白质增加和减少的代表性 KEGG 途径。(C) 箱线图显示肿瘤与 SCLC 相关蛋白的配对 NAT 之间的变化,并由人类蛋白图谱注释了潜在的临床实用性。(D) 散点图显示两种 SCLC 组织学类型(CSCLC、组合 SCLC)中肿瘤和配对 NAT 之间蛋白质变化的比较。(E) GO分析。(F) 火山图显示肿瘤和配对 NAT 之间差异表达的磷酸位点。(G) 散点图描绘了磷酸位点及其相应蛋白质之间丰度变化的比较。(H) 通过 PTM 特征富集分析 (PTM-SEA) 评估肿瘤和配对 NAT 之间的显著激酶特征。(I) 该图显示根据其底物的磷酸化水平和相应的激酶激活位点推断出活性增加的激酶。(J) 点图显示激酶底物磷酸化的变化。
04
对已识别的与生存相关的蛋白质进行功能分析
他们进行了监督分析来识别预后生物标志物,并揭示了与患者生存相关的 16 种差异表达蛋白(图4A)。其中,HMGB3在肿瘤中表达升高,与较差的生存率相关,而CASP10表达降低,与较好的预后相关。通过对来自数据产生 TU-SCLC 队列和另一个独立 SCLC 队列的样本进行免疫组织化学(IHC) 分析进一步验证(图4B-D),证实了它们的潜力SCLC 的预后价值。
HMGB3属于高迁移率族超家族,与核小体结合,参与DNA复制、重组、修复和转录。在许多癌症中观察到 HMGB3 高表达,并且与耐药性和患者生存率低相关。由于 HMGB3 在 SCLC 中表达上调,他们推测 HMGB3 可能作为 SCLC 中的致癌驱动因素。事实上,H345 细胞中 HMGB3 的过度表达增加了细胞迁移,而 HMGB3 敲低则导致细胞迁移减少(图 4E-F)。他们在 H345 细胞中过表达 HMGB3,并进行染色质免疫沉淀测序 (ChIP-seq) 分析,以鉴定潜在的 HMGB3 靶基因。富集分析表明,HMGB3 结合的基因参与控制细胞迁移表型的途径,例如黏附连接、紧密连接和黏着斑。他们重点关注先前报道的 5 个促进癌细胞迁移的基因,包括CLDN10、 PKP2、 ITGB4、 VTN和LAMC2。与 ChIP-seq 结果一致,HMGB3 过表达导致这些基因在 mRNA 水平的表达增加(图 4 G)。因此,他们假设HMGB3可能调节CLDN10、 PKP2、ITGB4、 VTN和LAMC2的转录,从而HMGB3有助于SCLC细胞的高迁移率。正如预期的那样, CLDN10、 PKP2、ITGB4、 VTN或LAMC2的敲低显著减少了亲本和 HMGB3 过表达的 H345 细胞中的细胞迁移(图 4H)。这些结果表明,HMGB3 是一种有价值的预后生物标志物,并通过细胞连接相关基因的转录调节作为 SCLC 的致癌因子发挥作用(图 4 I)。
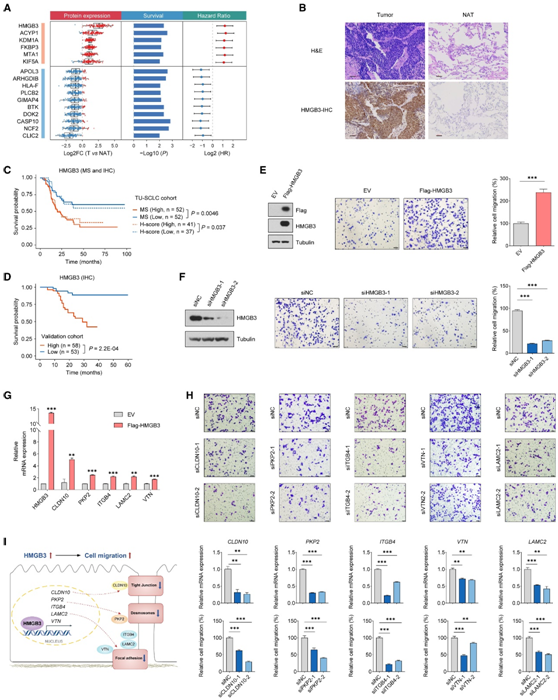
图4. 蛋白质组预后生物标志物的鉴定和验证。
(A) 代表 16 种候选预后蛋白的图表。(B) 肿瘤和配对 NAT 上 HMGB3 的代表性苏木精和伊红(H&E) 和免疫组织化学 (IHC) 染色图像。(C) 基于 HMGB3 蛋白质组丰度或免疫染色评分(对数秩检验)的总体生存 Kaplan-Meier 曲线。(D) 基于独立 SCLC 队列中 HMGB3 免疫染色评分的 Kaplan-Meier 总生存曲线。(E-F) HMGB3 过表达或敲低对 SCLC 细胞迁移的影响。(G) 与载体对照相比,过表达 HMGB3 的 H345 细胞中代表性 HMGB3 靶基因的实时 qPCR 验证。(H) CLDN10、PKP2、ITGB4、VTN 和 LAMC2 敲低对 SCLC 细胞迁移的影响。(I)HMGB3 在 SCLC 细胞迁移中的作用的示意性工作模型。
05
TU-SCLC 队列的免疫状况
他们使用估计免疫评分来评估免疫细胞浸润水平,这些结果表明免疫细胞浸润与性别、吸烟、TNM分期和患者生存显著相关。基于肿瘤和 NAT 的 xCell 衍生细胞类型富集评分进行无监督聚类,确定了三种免疫聚类,包括热肿瘤富集、冷肿瘤富集和 NAT 富集亚型(图 5 A)。大多数肿瘤属于免疫冷亚型,且预后较差(图5B)。有趣的是,免疫冷肿瘤表现出较高的 NE 评分(图 5 C),并且免疫评分与 NE 评分之间存在显著的负相关,这进一步表明免疫冷肿瘤与 NE 表型之间的联系。此外,免疫热亚型的特点是细胞毒性免疫细胞[CD8+T 细胞、自然杀伤细胞、活化树突状细胞(DC)和M1巨噬细胞]和免疫抑制细胞(调节性 T 细胞和 M2巨噬细胞),并显示出多种免疫相关途径的上调,包括干扰素-γ反应、抗原加工和呈递以及自然杀伤细胞介导的细胞毒性(图5A)。T细胞标志物(CD3D、CD8A)、MHC分子、刺激和抑制性免疫调节蛋白特征、细胞溶解活性,以及免疫检查点分子(CTLA4、PD-L1、PD-1和IDO1)在免疫热肿瘤中的表达与免疫冷肿瘤相比也上调(图5A)。因此,他们推测免疫疗法可能有利于免疫热亚型的SCLC人群。
值得注意的是,他们在免疫热肿瘤中观察到ZFHX3突变的显著富集(图5D)。与ZFHX3- WT肿瘤相比,具有ZFHX3突变的肿瘤表现出更高的免疫评分和TMB(图5E-F),表明ZFHX3突变与肿瘤免疫原性升高相关。ZFHX3突变患者似乎比ZFHX3 -WT 患者有更好的生存率。ZFHX3突变体样品中显著上调的蛋白质在免疫途径中富集,包括对干扰素 γ 的反应以及抗原加工和呈递。一致地,MHC分子、刺激性和抑制性免疫调节剂以及xcell来源的CD4+、CD8+ T细胞、DC和M1巨噬细胞特征在zfhx3突变肿瘤中上调(图5G-H),而与zfhx3突变相关的NAT在这些免疫细胞中没有统计学差异。因此,他们将ZFHX3突变与免疫激活行为联系起来,并推测ZFHX3突变可能是免疫治疗的预测性生物标志物。主要病理反应(Major pathologic response, MPR)用于评价新辅助免疫治疗疗效的病理反应,其定义为治疗后肿瘤残留不超过10%。其中5例为MPR, 7例为非MPR。WES结果提示3例患者存在ZFHX3突变,值得注意的是,3例ZFHX3突变患者(100%)均属于MPR,而只有2例WT患者(22%)为MPR(图5I-J)。同样,在这12例临床试验患者中,zfhx3突变组的残留肿瘤细胞数量明显低于ZFHX3-WT组(图5K),表明ZFHX3突变的患者可能从免疫治疗中受益更多。
为了进一步探索免疫冷肿瘤的潜在决定因素,他们使用免疫评分和蛋白质组数据进行了相关性分析。正如预期的那样,免疫相关途径显示出正相关,而 DNA 复制、细胞周期和 DNA 损伤修复途径与免疫评分呈负相关(图5L)。由于癌细胞中DDR的缺乏对肿瘤免疫原性至关重要,而SCLC细胞由于极高的基因组不稳定性和复制应激而特别依赖高DDR活性来生存,因此他们推测DDR活性升高可能导致SCLC的免疫抑制反应,特异性DNA修复途径评分与免疫评分之间存在负相关。DNA修复蛋白(PARP1、MSH2、MSH6、XRCC1、XRCC5、XRCC6)和DNA损伤检查点(CHEK1、WEE1、AURKA/B)在免疫冷肿瘤中的蛋白丰度和磷酸化水平也有所增加。此外,胞质DNA传感通路与免疫评分呈强正相关(图5L-M),提示胞质双链DNA激活的先天免疫关键通路cGAS-STING通路可能与免疫浸润有关。STING通路活性和下游促炎趋化因子CCL5和CXCL10的表达与免疫特征显著相关(图5N)。
总之,这些蛋白质组数据表明,DDR 活性升高可能通过减弱 SCLC 中 cGAS-STING 通路的激活而导致免疫抑制。
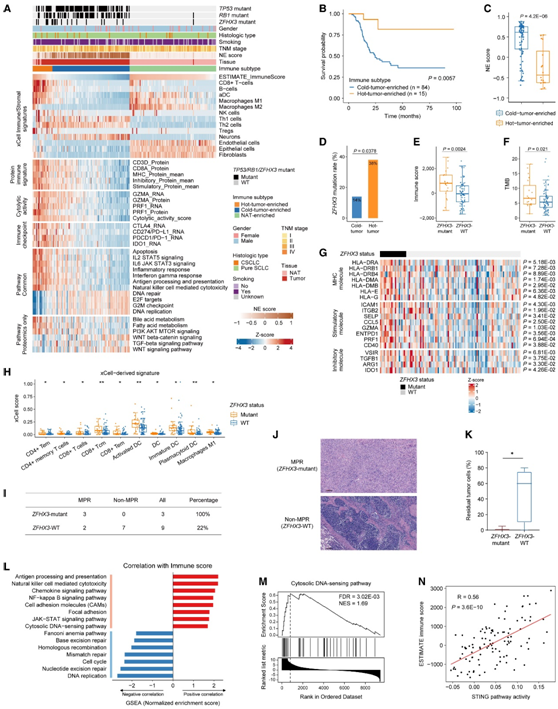
图5. TU-SCLC 队列的免疫状况。
(A) 热图显示了基于 xCell 衍生细胞特征的三个免疫聚类。(B) 基于免疫亚型的总体生存 Kaplan-Meier 曲线。(C) 冷肿瘤富集亚型和热肿瘤富集亚型之间的 NE 评分比较。(D)免疫冷肿瘤和免疫热肿瘤之间ZFHX3突变率的比较。(E-F) ZFHX3-突变体和ZFHX3 -WT肿瘤之间的免疫评分和TMB的比较。(G)说明ZFHX3-突变体和ZFHX3 -WT肿瘤之间涉及MHC分子、刺激性和抑制性免疫调节剂的差异表达蛋白的热图。(H) ZFHX3-突变体和ZFHX3 -WT 肿瘤之间xCell 衍生的免疫细胞特征的比较。(I) 正在进行的临床试验中观察到的ZFHX3突变与免疫治疗反应之间的关联。(J) 具有ZFHX3突变的患者的 MPR 和接受新辅助免疫治疗联合化疗的无ZFHX3突变的患者的非 MPR 的代表性 H&E 图像。(K)来自临床试验患者的ZFHX3突变体和ZFHX3 -WT组之间残留肿瘤细胞的比较。(L) 条形图显示与免疫评分相关(红色)或反相关(蓝色)的KEGG 通路的归一化富集评分。(M) 与免疫评分相关的胞质 DNA 传感通路的 GSEA 图。(N) 散点图显免疫评分与STING 通路活性的相关性。
06
具有独特生物学特征的多组学亚型
他们通过整合来自 107 个 SCLC 肿瘤的 mRNA、蛋白质和磷酸化数据,应用基于非负矩阵分解 (NMF) 的无监督聚类,并将它们分为四个聚类(图6A)。nmf1亚型主要富集于细胞周期、DNA损伤、染色质组织和表观遗传调控途径,表明该亚群具有高增殖率。nmf2亚型的特征蛋白或磷酸化位点较少,但TMB最高(图6B)。nmf3亚型在蛋白水平上与ECM受体相互作用、ECM组织和黏着斑途径相关。值得注意的是,磷酸化蛋白质组学数据显示,nmf3亚型中受体酪氨酸激酶(rtk)的信号活性增加。最后,nf4亚型以MYC靶点的高表达和RNA代谢途径的富集为特征,并且具有较高的染色体不稳定性(CIN)、干性评分和mrna -蛋白相关性(图6A-B)。同样,亚型特异性途径富集分析也显示了四种亚型之间不同的分子特征。
每种亚型均显示出不同的 CNA 特征。TP53、RB1和FAT1的缺失在nmf1和nmf2亚型中显著富集,而ZFHX3缺失主要局限于nmf1亚型。相比之下,癌基因 AURKA和GNAS扩增优先与 nmf4 亚型相关(图 6 C)。他们根据 mRNA 表达检查了四种亚型中转录因子 ANPY(ASCL1、NEUROD1、POU2F3和YAP1)的表达模式,发现了明显的分离(图 6D-E)。nmf1亚型显示ASCL1、NEUROD1或两者的高表达。POU2F3 的高表达主要限于 nmf4 亚型,并且与 ASCL1 和 NEUROD1 相互排斥。尽管YAP1在 nmf3 和 nmf4 亚型中的表达相对较高,但与其他三种转录因子相比,YAP1 的表达非常低。此外,他们观察到 nmf1 亚型的 NE 得分最高(图 6 F),同时伴有 NE 标记 SYP、CHGA、NCAM1 和 INSM1 的高蛋白表达(图 6 D)。nmf3 亚型显示出最高的 EMT 评分(图 6 G),这与 SCLC 转移和化疗耐药相关。
MYC的扩增显示这些亚型之间没有统计学上的显著差异,而 MYC mRNA 表达在 nmf4 亚型中显示出显著上调(图6H)。此外,SCLC 肿瘤和 NAT 中 MYC 蛋白的IHC染色证实了这些差异(图 6I-J)。磷酸蛋白质组学数据显示,在 nmf4 亚型中,MYC S62、S347、S348 和 S161 磷酸位点也上调(图 6K)。其中,MYC S62是研究最多的磷酸位点,它可以促进MYC的稳定并增强MYC活性。这些结果表明 MYC 在 nmf4 亚型中具有活跃的致癌作用。与 NAT 样本相比,MYC 表达仅在 nmf4 亚型肿瘤中在RNA、蛋白质和磷酸化水平上上调,而在其他亚型中下调(图 6L-M)。这些研究结果表明,MYC 表达(而不是MYC基因扩增)可以更好地指示 MYC 状态,并且可能是定义具有低 NE 特性和高 POU2F3 表达的 SCLC 子集的更有价值的标记。
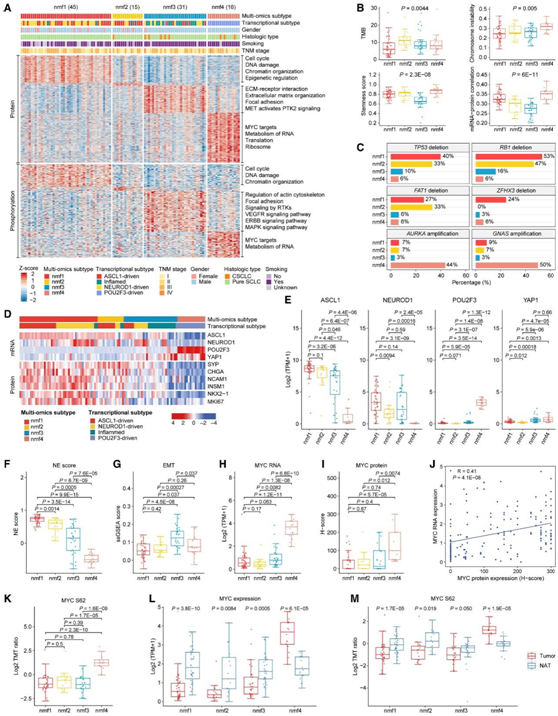
图6. TU-SCLC队列的多组学分类及相应的生物学特征。
(A) SCLC 肿瘤的综合多组学分类为四种 NMF 衍生亚型。(B) 跨多组学亚型的 TMB、染色体不稳定性 (CIN)、干性评分和 mRNA-蛋白质相关性的比较。(C) 每个亚型具有 CNA 的特定基因的百分比。(D) 热图显示多组学亚型中ANPY的相对 mRNA 表达、神经内分泌标记物、甲状腺转录因子 1 (TTF-1/NKX2-1) 和增殖指数标记物 Ki-67/MKI67 的蛋白表达。(E) 多组学亚型之间 ANPY 表达的比较。(F-G) 比较从不同亚型的蛋白质组数据推断的 NE 分数和 EMT 分数。(H-I) 通过 IHC 检测比较不同亚型的 MYC mRNA 丰度和蛋白质丰度。(J) 散点图显示 MYC mRNA 与蛋白质丰度(H 分数)的 Spearman 相关性。(K) 不同亚型的 MYC S62 丰度比较。(L-M) 分别比较每个亚型中肿瘤和配对 NAT 之间的 MYC mRNA和 MYC S62磷酸位点丰度。
07
SCLC 亚型特异性治疗策略
基于多组学亚型的分子特征,他们探索了 SCLC 亚型特异性的治疗弱点。nmf1 亚型与高增殖、 E2F活性以及复制应激相关。磷酸化蛋白质组数据表明 RTK 信号传导活性在 nmf3 亚型中显著上调(图 6 A)。安罗替尼(anlotinib)是一种多靶点酪氨酸激酶抑制剂,以其抗血管生成和抗肿瘤活性而闻名,它可以改善 SCLC 患者的无进展生存期 (PFS) 和 OS。
他们检测了RTK 抑制剂安罗替尼在包括 SC022、SC224 和 DMS114 在内的多个 nmf3 PDX/CDX 模型中的治疗效果。在所有三种 nmf3 肿瘤模型中均观察到对肿瘤生长的强烈抑制(图7A-C),而在 nmf1 肿瘤模型(H146 和 SC234)中未观察到明显的抑制(图 7J-L)。他们分析了 AURK 抑制剂(alisertib、barasertib 和 AMG-900)对 nmf4 SCLC 细胞系(H82 和 H446)以及 PDX/CDX 模型(SC222 和 H446)的作用。与 nmf1 亚型 (H146) 相比,nmf4 细胞系对 alisertib、barasertib 和 AMG-900 治疗更敏感(图7D -7F)。此外,alisertib治疗显著抑制nmf4 PDX/CDX模型中的肿瘤生长(图7G-I),而在nmf1肿瘤模型(H146和SC234)中几乎没有引起任何抑制(图7J-L)。
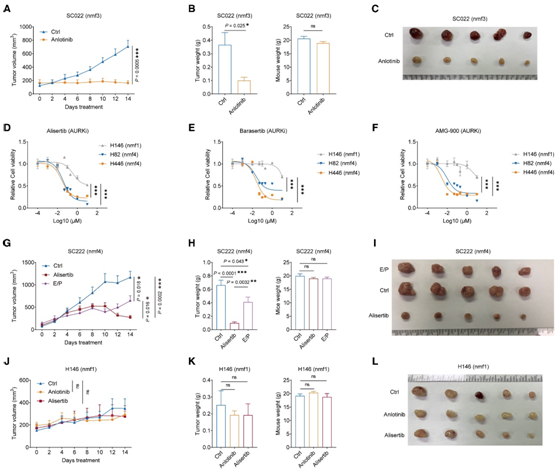
图7. SCLC 亚型特异性治疗策略。
(A) 用安罗替尼或载体治疗的 SC022 荷瘤小鼠中的肿瘤生长。(B) 对用安罗替尼或载体治疗的 SC022 PDX 模型的肿瘤重量(左)和小鼠重量(右)进行统计分析。(C) 来自用安罗替尼或载体处理的 SC022 PDX 模型的肿瘤。(D–F) H146、H82 和 H446 细胞的相对活力。(G) 用 E/P、alisertib 或载体治疗的 SC222 荷瘤小鼠中的肿瘤生长。(H) 对用 E/P、alisertib 或载体处理的 SC222 PDX 模型的肿瘤重量(左)和小鼠重量(右)进行统计分析。(I) 来自用 E/P、alisertib 或载体处理的 SC222 PDX 模型的肿瘤。(J) 用安罗替尼、alisertib 或载体处理的 H146 荷瘤小鼠中的肿瘤生长。(K) 对用安罗替尼、alisertib 或载体处理的 H146 CDX 模型的肿瘤重量(左)和小鼠重量(右)进行统计分析。(L) 来自用安罗替尼、alisertib 或载体处理的 H146 CDX 模型的肿瘤。
+ + + + + + + + + + +
结 论
本项研究使用 112 名接受手术切除的初治患者的配对肿瘤和邻近肺组织对小细胞肺癌进行了全面的蛋白质组学表征。综合多组学分析阐明了遗传畸变下游的癌症生物学,并强调了FAT1突变、RB1缺失和染色体 5q丢失的致癌作用。确定了预后生物标志物 HMGB3 。HMGB3 的过度表达通过细胞连接相关基因的转录调节促进 SCLC 细胞迁移。免疫特征分析揭示了ZFHX3突变与高免疫浸润之间的关联,并强调了通过抑制 cGAS-STING 途径提高DNA 损伤反应活性的潜在免疫抑制作用。多组学聚类确定了四种具有亚型特异性治疗靶点的亚型。基于细胞系和源自患者的异种移植药物测试验证了多组学亚型预测的特定治疗反应。这项研究为更好地了解 SCLC 生物学和改善临床实践提供了宝贵的资源和见解。
+ + + + +



