

 English
English文献解读|Cancer Cell(50.3):综合分子和空间分析揭示了原位和侵袭性肢端黑色素瘤的进化动力学和肿瘤免疫相互作用
✦ +
+
论文ID
原名:Integrative molecular and spatial analysis reveals evolutionary dynamics and tumor-immune interplay of in situ and invasive acral melanoma
译名:综合分子和空间分析揭示了原位和侵袭性肢端黑色素瘤的进化动力学和肿瘤免疫相互作用
期刊:Cancer Cell
影响因子:50.3
发表时间:2024.05.09
DOI号:10.1016/j.ccell.2024.04.012
背 景
肢端黑色素瘤(AM)源于手掌、脚底和指甲的黑色素细胞,是亚洲人黑色素瘤的主要亚型。AM通常在晚期诊断,经常发生转移,预后不佳。早期发现和预防AM可显著改善患者预后。目前的研究主要集中在晚期侵袭性AM,在肢端黑色素瘤 (AM) 中,从原位AM (AMis) 发展为侵袭性 AM (iAM) 会导致生存率显著降低,目前对早期AM的了解仍然非常有限。
实验设计

结 果
01

发现队列和基因组景观
研究团队组建了一个由 287 名患者组成的 AM 队列,其中包含一个由 147 名患者组成的核心发现队列和两个由 140 名患者组成的验证队列。其中,分别有146名患者和141名患者诊断为AMis和iAM。该发现队列包含 56 名 AMis 患者和 91 名 iAM 患者(图 1 A,图S1A-D)。AMis 患者比 iAM 患者更年轻,分别有 15% 和 47% 的患者出现溃疡和附件受累。值得注意的是,许多 iAM 肿瘤在表皮中含有恶性细胞,表现为 AMis 病变,这些肿瘤也可称为同步 AMis-iAM。同步 AMis-iAM 的这种双重表型使其成为研究患者体内 AM 侵袭进化动力学的理想模型。他们应用激光捕获显微切割的多区域测序(LCM)从同一患者中分离出匹配的高纯度肿瘤样本(图 1 B-C),包括来自 12 名患者的 12 个 Syn_AMis 和 11 个 Syn_iAM 区域。
全外显子组测序(WES)的平均覆盖率为 313×,能够实现准确的突变识别和克隆组成重建(图 S1 E)。本项研究队列的肿瘤突变负荷(TMB)与之前的AM队列相当(图1D)。AM的广泛拷贝数改变(CNA)谱(图1E,图S1F-H)和突变特征与以前的AM组相似,但与皮肤黑色素瘤(CM)和葡萄膜黑色素瘤(UM)组不同。例如,最近报道的22q11.21扩增代表着预后很差(图S1H)。

图1. 研究策略和队列信息。
(A) 研究方案。(b) 代表性 H&E 染色。(C) 每个 AJCC 阶段不同肢端黑色素瘤 (AM) 队列的组成。(D) 黑色素瘤队列中的肿瘤突变负荷 (TMB)。(E) 黑色素瘤队列中的改变频率。

图S1. AM的队列组成和基因组特征。
(A)发现队列、扩展队列和验证队列中AM患者的组成。(B)发现队列中每个测序平台涉及的患者和样本数量。(C)参与WES和bulk RNA-seq的样品。(D) amis和iAM患者临床特征饼状图。(E)对比WES样本测序覆盖率的箱线图。(F) GISTIC2在整个队列中确定的病灶区域。(G) AM中选定信号通路中基因频率改变的比较。(H)按22q11.21扩增状态分层的AM患者总生存期(OS)。(I) SigProfilerExtractor分析。(J)在AM中发现的从头特征。
02
AMis 和 iAM 的基因组比较确定了侵袭偏好的驱动因子
为了确定驱动AM侵袭的潜在事件,他们比较了amis和iAM样本之间的基因组景观(图2A)。TMB具有可比性(图2B),amis和iAM中均没有单个突变显著富集。基因组合筛选发现,在iAM样本中,四个驱动基因(NRAS、KRAS、NF1或KIT)的突变频率显著较高(图2C)。他们将这四种基因称为AM的“侵袭偏好驱动因子”。诱导热点突变(包括NRASQ61K、NRASQ61R、KRASG12D和KITL576P)和敲除NF1可增强AM细胞系LM- MEL -45的侵袭(图2D),支持侵袭偏好驱动因子的功能作用。
亚克隆突变(即某个肿瘤中所有肿瘤细胞不共有的突变)的百分比可以评估肿瘤内异质性(ITH)的程度。有趣的是,iAM 样本的亚克隆突变比例高于 AMis 样本(图 2 E),这表明高水平的 ITH 与侵袭性相关。接下来他们用三个独立指标量化基因组不稳定性水平,包括加权基因组完整性指数 (wGII) 评分、染色体拷贝数异质性评分 (CNH-DNA) 和染色体不稳定性评分 (CIN-RNA)。正如预期的那样,iAM表现出明显高于amis的基因组不稳定性(图2F)。这些结果表明,高度的基因组不稳定性可能促进肿瘤的发生。
AM 的附件受累(定义为小汗腺管中存在恶性黑色素细胞)是 iAM 中与 AMis 相比唯一显著富集的变量(图2G)。虽然 TMB 可比,但无论是所有患者还是 AMis 患者,附件受累的 AM 中侵袭性驱动因子的突变频率都显著更高(图 2 H)。这些结果暗示获得侵袭性驱动突变的 AMis 肿瘤细胞可能具有更高的增殖优势,并受到正向选择和克隆扩增 (CE)。附件受累可能作为区分具有侵袭潜力的 AMis 肿瘤和惰性肿瘤的病理预测因素。对139 名 AMis 患者的回顾性分析显示,附件受累与 AMis 的总体生存期 (OS)(图 2 I)和无进展生存期 (PFS)(图2J)缩短相关。

图2. AMis 和 iAM 之间的基因组比较。
(A) 体细胞改变景观。(B-C) TMB和侵袭偏好驱动因子的比较。(D) 具有驱动基因诱导或敲除的 LM-MEL-45 AM 细胞系的 Transwell 侵袭测定。(E-F) 亚克隆突变和 wGII 评分的比较。(G) 有或无附件受累的患者频率。(H) 所有患者(左)和 AMis 患者(右)中有或没有附件受累的侵袭偏好驱动因子的突变频率。(I-J) 按附件受累状态分层的 AMis 的总生存期(OS)和无进展生存期(PFS)。
03
同步 AMis-iAM 揭示了侵袭的早期单克隆播种模式
为了研究患者内侵袭和区域扩张的进化动态,他们通过 LCM 分析了同步 AMis-iAM(图 3 A)。首先比较了三种不同类型病变的基因组景观,包括纯 AMis、Syn_AMis 和 Syn_iAM(图 1 B)。令人惊讶的是,在 5 名表现出侵袭偏好驱动因子的患者中,突变在配对的 Syn-AMis 和 Syn-iAM 共存,但在纯 AMis 中未检测到任何突变(图 3 B)。该结果表明侵袭偏好驱动因子是在侵袭之前获得的,进一步证实了它们在 AM 侵袭中的驱动作用。与其他病变相比,Syn_iAM 显示出明显更高的亚克隆突变百分比,但 TMB 相当,导致 ITH 更高(图 3 C-D)。他们还观察到 Syn_iAM 病变中的基因组不稳定性水平明显更高,如 wGII 和 CNH-DNA 评分升高所示(图3E-F)。这些结果证明了基因组不稳定性和 ITH 导致 AM 入侵。
对同一患者的匹配样本进行系统发育分析,发现普遍存在短树干和长分枝的模式,表明AM中ITH的程度很高。以树干长度为代表的共有突变百分率在AM中显著低于CM(图3G)。这一结果表明Syn_iAM与Syn_AMis的遗传分化相对较早,这与其预后较CM差的情况相一致。这种普遍存在的早期播种模式也不同于其他癌症的可变树结构,表明AM具有独特的侵袭过程。接下来他们推断了侵袭的起源和时间。Jaccard相似性指数(JSI)可以区分单克隆播种(JSI < 0.3)和多克隆播种(JSI≥0.3)。他们在所有患者中观察到普遍存在的单克隆播种模式(图3I)。与此一致的是,在Syn_AMis和Syn_iAM中,侵袭偏好驱动因子的突变大多是克隆性的(图3J)。患有多个 Syn_AMis 病变的患者可以提供有关 Syn_AMis 和 Syn_iAM 之间遗传关系的更多信息(图 3 K)。在患者 AM18 中,系统发育分析显示 Syn_iAM 起源于邻近的 Syn_AMis_2,而不是遥远的 Syn_AMis_1。值得注意的是,NRASQ61K突变是 Syn_AMis_2 和 Syn_iAM 共有的少数突变之一,但在 Syn_AMis_1 中不存在。这一结果表明Syn_AMis在获得NRASQ61K后很快就侵入真皮层,支持其驱动侵袭的作用。三个区域的非共有突变的相似突变谱表明存在共有诱变背景(图 3K)。
为了探究区域扩张的进化模式,他们通过癌细胞分数 (CCF) 分析推断了克隆组成(图 3 L)。成对 CCF 分布显示,81.8% (9/11) 的 Syn_AMis 病变呈现克隆峰,与 44.4% (4/9) 的 Syn_iAM 病变形成鲜明对比(图 3 M)。因此,Syn_AMis 倾向于表现出更均质的组成,这与前面提到的观察结果一致,即具有附件参与的 AMis(即高侵袭潜力)表现出更多的克隆突变。在 Syn_iAM 中,他们描述了区域扩张的两种不同的进化模式:(1) CE,其中侵袭性肿瘤保持克隆;(2) 亚克隆多样化 (SD),其中肿瘤获得不同的亚克隆(图 3 L)。分层为 SD 模式的患者表现出较高程度的 ITH,且不受肿瘤纯度的影响(图 3N-O)。鉴于高 ITH 可能会产生亚克隆驱动因子,他们推测 SD 过程促进了驱动因子的获取,从而促进了区域扩张(图 3 P)。

图3. AM入侵的进化动力学。
(A-B)激光捕获显微切割、LCM、区域的采样信息和突变景观。(C–F) 比较肿瘤突变负荷、TMB、亚克隆突变、wGII和 CNH-DNA评分的箱线图。(G) AM19 的系统发育树。(H) 共有变体的比较。(I)具有两个或多个区域的患者的杰卡德相似指数(JSI)。(J) 侵袭偏好驱动因子的癌细胞分数 (CCF) 图。(K) AM18 的系统发育树(左)、示意图(中)和突变谱(右)。(L) 克隆扩增 (CE) 和亚克隆多样化 (SD) 的代表性 CCF 图。(M) 饼图显示 CE 和 SD 的比例。(N-O)肿瘤纯度和TMB的比较。(P) AM 入侵过程中克隆动力学的示意图。
04
综合分析确定了三种分子亚型
他们使用bulk RNA-seq 数据对 81 个 AM 样本进行无监督层次聚类,确定了三种不同的分子亚型,分别命名为 C1、C2 和 C3(图 4 A)。亚型 C1 显示角蛋白基因表达升高、皮肤发育和角化通路和丝裂原活化蛋白激酶 (MAPK) 活性低(图 4 B),因此将其称为“角蛋白”亚型。亚型 C2 显示染色质重塑基因(KMT2A和KMT2C)、DNA 和组蛋白甲基化途径上调,表明存在“染色质重塑”表型。亚型 C3 表现出 EMT 相关基因 (TWIST2和VIM)的上调、ECM 组织和巨噬细胞浸润途径以及高增殖活性,共同提出“增殖”表型(图4A-B)。C3 亚型表现出最高的 CIN-RNA、wGII 和 CNH-DNA 评分,表明基因组高度不稳定(图 4 C)。一致地,C3 亚型显示出最高比例的亚克隆突变(图 4D)。这些结果从基因组角度共同解释了C3亚型的恶性特征。正如预期的那样,三种亚型表现出不同的 PFS 和 OS,其中 C3 亚型的预后最差(图 4E)。
与 C1 和 C2(AMis 和 iAM 的均匀混合物)相比,C3 亚型高度富集 iAM (27/28)(图 4 A),表明 iAM 的独特子集。与C1和C2中的iAM(以下称为非C3 iAM)相比,C3 iAM表现出缩短的PFS和明显的恶性表型(图4F)。重要的是,C3 iAM 表现出明显更多的亚克隆突变(图 4 G)。 81% 的 C3 iAM 对应于 SD 模式,相比之下,44% 的非 C3 iAM 对应于 SD 模式(图 4 H)。这一结果部分反映了 C3 iAM 的不良预后。值得注意的是,根据 Breslow 厚度和溃疡情况确定,两组之间的肿瘤 T 分期没有显著差异(图 4 I),这表明 AM 的浸润深度可能与其进展能力并不一定相关。多变量分析证实 AM 亚型是一个独立的预后因素(图 4 I)。
随即,他们观察到三种分子亚型的共有 TME 表型(图 4 J)。C1亚型显示出最低水平的免疫浸润和促肿瘤细胞因子,命名为“低免疫”。C2亚型显示出最高丰度的B细胞和相对较高的T细胞细胞毒性,命名为“免疫激活”。C3 亚型显示出最高水平的免疫浸润、肿瘤相关巨噬细胞 (TAM) 和 EMT。C3 中的 TAM 主要是免疫抑制性巨噬细胞(图 4 K),高表达CD163、APOE、C1QC(图 4 L)、以及VSIG4和CSF1R等治疗靶点,这些结果共同提出了 C3 的“免疫抑制”表型。
TAM 评分与基因组不稳定指数相关,包括 wGII(图 4 M)和 CNH-DNA 评分以及亚克隆突变(图 4 N)。此外,TAM 评分与 EMT 评分(图 4 O)以及队列中的不良预后相关(图 4 P-Q)。这些结果共同表明,TAM 浸润可能会促进肿瘤 EMT 和 C3 亚型的基因组不稳定,并导致不良预后。

图4. AM 的分子亚型和 TME。
(A) 从大量 RNA-seq 数据推断出的三种分子亚型。(B) 基因本体 (GO) 分析。(C-D) CIN-RNA、wGII 和 CNH-DNA 评分以及亚克隆突变的比较。(E-F) 基于分子亚型的所有患者和 iAM 患者的无进展生存期 (PFS)。(G) 亚克隆突变的比较。(H) iAM 患者分配至亚克隆多样化 (SD) 或克隆扩增 (CE) 的比例。(H) iAM 患者分配至亚克隆多样化 (SD) 或克隆扩增 (CE) 的比例。(J) 肿瘤微环境(TME)特征的比较。(K) C3 AM 的 TAM 组成。(L) 免疫抑制 TAM 标记的表达。(M–O) TAM 与亚克隆突变、wGII或 EMT 评分之间的相关性。(P-Q) 按 TAM 评分分层的 AM 患者的总生存率和 PFS。
05
scRNA-seq 揭示 APOE + /CD163 +巨噬细胞亚群
为了在单细胞水平上探究肿瘤和 TME 异质性,他们对 8 个 AMis 和 16 个 iAM 肿瘤进行了单细胞转录组分析(scRNA-seq)。总共收获了 223023 个细胞,并将其分为 13 个主要细胞群(图 5 A)。对每个亚型的特征基因进行的分析成功鉴定了所有三种亚型(图 5 B)。恶性肿瘤细胞可分为8种细胞状态,包括黑素细胞、EMT、IFN反应、抗原呈递、循环、RNA加工和两种应激相关状态(图5C-D),C3 亚型中的肿瘤细胞表现出最高的 EMT 分数(图 5 E)。
他们确定了巨噬细胞的五个子集,包括 Mph_APOE_CD163、Mph_APOE、Mph_CD163、Mph_ISG15 和 Mph_TIMP1(图 5 F)。极化分析表明Mph_APOE_CD163、Mph_CD163和Mph_APOE都是免疫抑制性TAM,因此统称为APOE+/CD163+TAM。APOE+/CD163+TAM在 C3 亚型中显著富集(图 5 G),并与 EMT 评分呈正相关(图 5 H),验证了在总体水平上的观察结果。配体-受体分析表明巨噬细胞表现出最丰富的细胞间相互作用。在 APOE+ /CD163+巨噬细胞内观察到高水平的募集和相互作用(图 5 I)。APOE + /CD163 +巨噬细胞可能通过 EMT 相关信号通路促进肿瘤细胞表型转变,包括TGFB1-TGFBR2、TNF-TNFRSF1B和IGF1-IGF1R(图5J)。多色免疫组织化学(mIHC)验证了APOE +巨噬细胞(Mph_APOE)、CD163 +巨噬细胞(Mph_CD163)和双阳性巨噬细胞(Mph_APOE_CD163)的存在(图5K)。

图5. 单细胞分辨率下 AM 的 TME。
(A) 细胞注释。(B) 基于 scRNA-seq 对 24 个肿瘤进行亚型分析。(C) 肿瘤细胞的细胞状态。(D) 富集了每个细胞状态的功能通路。(E) 上皮-间质转化 (EMT) 三种亚型的评分。(F) 肿瘤相关巨噬细胞(TAM),由聚类身份(左)和标记基因表达(右)标记。(G) APOE + /CD163 +巨噬细胞比例的比较。(H) APOE + /CD163 +巨噬细胞比例与患者 EMT 评分之间的 Sperman 相关性。(I)基于CellPhoneDB的配体-受体分析。(J)基于NicheNet的配体-受体分析。(K) 代表性 mIHC 图像显示 APOE +(左)、CD163 +(中)和 APOE + CD163 +巨噬细胞(右)。
06
空间转录组分析
为了研究 APOE + /CD163 + TAM 是否与 EMThigh肿瘤细胞在空间水平上相互作用,他们对 10 个 AM 样本进行了空间转录组学分析 (ST)(图6A)。多个表皮层的结构,包括基底层、棘层和颗粒层,以及小汗腺和血管的皮下结构都可以清晰地可视化(图6A)。肿瘤区域可以通过黑色素瘤特征基因的高表达来识别,如MLANA、PMEL、TYPR和DCT(图 6 B)。例如,在 C3 iAM 患者 AM140 中,肿瘤区域由 EMT高肿瘤细胞组成,其特征是CDH2表达升高,并由 APOE + /CD163 + TAM 高度浸润。相比之下,在非 C3 患者 AM147 中无法发现这种共存。
单细胞分割后,根据Voronoi图进行定量分析。通过使用CytoSPACE将ST数据与scRNA-seq整合来确定细胞身份。定位到单细胞肿瘤EMT状态的细胞定义为EMT肿瘤细胞,而非EMT肿瘤细胞定义为那些定位到其他肿瘤状态的细胞(图6C)。正如预期的那样,C3患者EMT肿瘤细胞比例明显更高,APOE+/CD163+巨噬细胞浸润水平也明显更高(图6D)。在C3患者中,与非EMT细胞相比,APOE+/CD163+巨噬细胞更靠近EMT肿瘤细胞(图6E)。CellChat分析显示,APOE+/CD163+巨噬细胞作为发送者(配体)和接收者(受体)均表现出较高的活性,证实了细胞间的高水平相互作用(图6F)。单细胞分析表明,IGF1-IGF1R可能介导APOE+/CD163+巨噬细胞与EMT肿瘤细胞之间的相互作用。与这一发现一致,与其他免疫细胞相比,APOE+/CD163+巨噬细胞表现出最高水平的胰岛素生长因子(IGF)信号网络(图6G)。这些空间数据揭示了APOE+/CD163+ tam与EMT肿瘤细胞之间的直接接触和密切相互作用。

图6. F5-CAF 与 HCC CSC之间的细胞相互作用。
(A) HCC界面区域的代表性复合 mIF 图像。(B) TME中 F5-CAF 配体受体(L-R)分析的示意图。(C) NicheNet 预测的 F5-CAF 和其他调节癌细胞的成纤维细胞亚群中配体的平均表达。(D) F5-CAF 表达的选定配体的空间特征图和具有干细胞特性的癌细胞的同源受体表达。(E) 不同位置的 L–R 共表达得分。
07
多重空间蛋白质组学分析验证 APOE + /CD163 + TAM 与 EMT 肿瘤细胞之间的相互作用
为了在蛋白质水平上进一步验证 APOE + /CD163 + TAM 与 EMT 肿瘤细胞之间的空间分布和相互作用,他们在 12 个 AM 样本中进行了 22 重索引联合检测 (CODEX)(图 7 A)。表皮以全细胞角蛋白 (pan-CK) 和 E-钙粘蛋白为标记。通过Melan-A的阳性表达来鉴定肿瘤细胞。肿瘤区域还观察到免疫细胞浸润,包括CD4 + T细胞(CD3e和CD4)、CD8+ T细胞(CD3e和CD8)、B细胞(CD20)、巨噬细胞(CD68)和树突状细胞(CD11c)。在邻近的正常区域,小汗腺以泛 CK 表达和导管结构为标志。通过直接接触小汗腺的肿瘤细胞可以清楚地识别附件受累。
C3患者中可以识别出EMT肿瘤区域,而非C3患者主要由非EMT肿瘤区域组成(图7B)。空间绘图显示 APOE+/CD163+TAM 与 EMT 肿瘤细胞之间呈正相关(图 7C)。在 C3 患者中观察到 IGF1 和 IGF1R 的表达水平显著较高(图 7D)。有趣的是,C3 患者 AM138 包含 EMThigh区域和 EMT低区域,从而能够进行患者内部比较(图 7 E)。APOE+/CD163+TAM在EMThigh区域高度积累(图 7 F)。EMT高区域中 IGF1 和 IGF1R 的表达也显著升高(图 7G)。这些结果证明APOE+/CD163 +TAM可能通过IGF1-IGF1R相互作用促进肿瘤细胞的EMT。
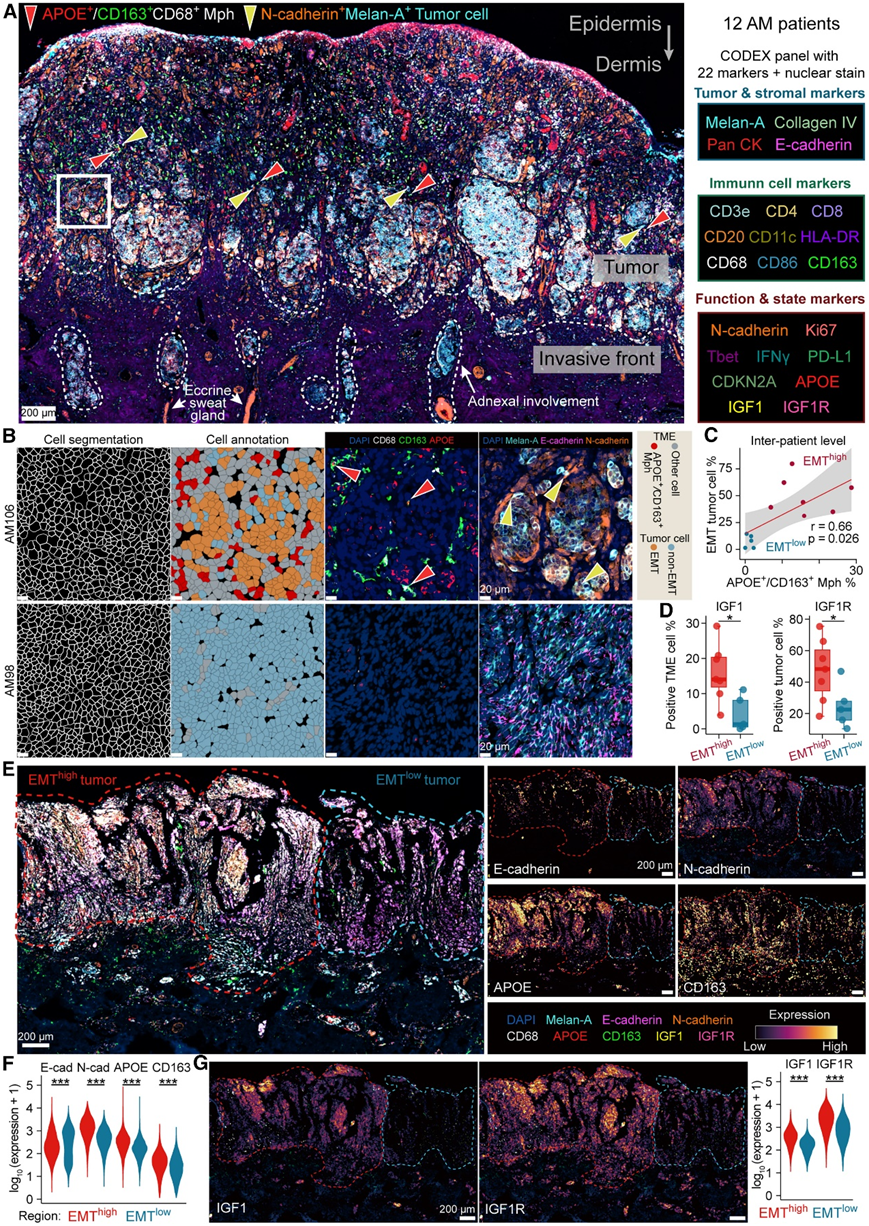
图7. 空间蛋白质组学分析。
(A) 具有代表性的 CODEX 图像。(B) 显示细胞分割、注释、APOE + /CD163 +巨噬细胞(红色箭头)和 EMT 肿瘤细胞(黄色箭头)的代表性图像。(C) APOE + /CD163 +巨噬细胞浸润与 EMT 肿瘤细胞之间的 Sperman 相关性。(D) IGF1 +(左)或 IGF1R +(右)细胞的比例。(E) AM138 的 CODEX 图像。(F) EMThigh区域和 EMTlow区域之间的分段细胞中的蛋白质表达水平。(G) IGF1 和 IGF1R 的空间图(左)和蛋白质表达水平(右)。
08
离体和体外试验表明 APOE+CD163+TAM 通过 IGF1-IGF1R 相互作用促进肿瘤 EMT
他们接下来进行了功能测定以探究 APOE+/CD163 +巨噬细胞与 EMT 肿瘤细胞之间的相互作用。从配对的外周血和肿瘤组织中分离的巨噬细胞的体外分析表明,APOE+CD163+巨噬细胞在肿瘤组织中显著富集(图 8 A)。为了研究 AM 肿瘤细胞是否可以直接诱导巨噬细胞的 APOE + CD163 +表型,他们从健康捐赠者中分离出单核细胞,并在体外将其与来自 LM-MEL-45 细胞系的条件培养基 (CM) 共培养。流式细胞术分析显示,APOE + CD163+巨噬细胞的比例从第 0 天的 1.2% 逐渐增加到第 14 天的 73.5%(图 8 B)。RNA-seq 分析也验证了 APOE + CD163 +巨噬细胞的时间依赖性诱导,如APOE、CD163、C1Q和IGF1等标志基因的表达持续升高(图8C)。基因集富集分析 (GSEA)显示,与对照组相比,第 14 天巨噬细胞中APOE + CD163 +特征的富集(图 8D )。巨噬细胞中APOE和CD163的这种升高的表达也可以在蛋白质水平上检测到(图 8E)。这些结果表明 AM 细胞可以诱导巨噬细胞的 APOE + CD163 +表型。
他们接下来探索 APOE + CD163 +巨噬细胞是否可以促进肿瘤细胞的 EMT。离体分析证实,C3 患者的 EMT 肿瘤细胞比例明显高于非 C3 患者(图 8 F)。体外与 APOE+CD163+巨噬细胞共培养后,LM-MEL-45 肿瘤细胞表达的间充质标志基因水平明显更高,包括CDH2、TWIST1/2、SNAI1/2等(图 8 G)。与单独的肿瘤细胞相比,共培养的肿瘤细胞中也富集了升高的 EMT 特征(图8H)。为探究APOE+CD163+巨噬细胞分泌IGF1的情况(图8I),ELISA分析显示,与APOE + CD163 +巨噬细胞共培养后,下室分泌的IGF1蛋白持续升高(图8J)。更重要的是,用IGF1抑制剂xentuzumab、IGF1R抑制剂linsitinib抑制该相互作用,或在肿瘤细胞中敲除IGF1R,均可显著降低N-cadherin的表达,使肿瘤细胞恢复圆形,E-cadherin重新表达(图8K-L)。综上所述,这些结果表明APOE+CD163+巨噬细胞可以通过IGF1-IGF1R相互作用促进肿瘤细胞的EMT。
在蛋白质水平上,他们对 96 名 AM 患者进行了 IHC 染色,其组织切片可在发现队列中获得(图 8 M)。值得注意的是,具有 APOE + CD163 +巨噬细胞的患者的五年 PFS 为 54.7%,远低于那些无APOE + CD163 +巨噬细胞的患者的 90.5%(图8N)。他们还在一个独立验证队列中对87例AM患者进行了免疫组化染色。同样,APOE和CD163双阳性染色与AM患者PFS缩短和OS缩短显著相关(图8O)。总而言之,这些结果表明 APOE 和 CD163 染色可以作为 AM 患者有希望的预后生物标志物。

图8. 功能测定。
(A) 流式细胞图。(B) 来自健康捐献者 PB 和 LM-MEL-45 肿瘤细胞系 CM 的单核细胞共培养系统。(C) APOE + CD163 +巨噬细胞的特征基因的 RNA 表达热图。 (D) 基于指定组之间 GSEA 分析的基因特征富集。(E) 显示巨噬细胞 APOE 和 CD163 表达的代表性图像。(F) 流式细胞图显示来自 C3 和非 C3 AM 样品的上皮间质转化 (EMT) 肿瘤细胞。(G) LM-MEL-45 细胞中 EMT 标记基因的 RNA 表达热图。(H) 基于 GSEA 分析丰富 EMT 特征。(I) scRNA-seq 数据中巨噬细胞子集的IGF1表达水平。(J) 共培养系统用于评估 APOE + CD163 +巨噬细胞对肿瘤细胞的影响。(K) 评估 (J) 中共培养系统中肿瘤细胞的 EMT 水平。(L) 代表性图像显示 LM-MEL-45 肿瘤细胞的 E-钙粘蛋白和 N-钙粘蛋白表达。(M) 阴性(无浸润)和阳性(浸润)患者中 APOE 和 CD163 的代表性 IHC 图像。(N-O) 通过 APOE 和 CD163 染色分层的发现队列和验证队列中 AM 患者的无进展生存期 (PFS) 和总生存期 (OS)。
+ + + + + + + + + + +
结 论
本项研究使用基因组学、单细胞转录组学以及空间转录组学和蛋白质组学报告了 147 个 AM 的综合分子和空间表征。从 AMis 到 iAM 的侵袭表现出早期的单克隆播种模式。 iAM 随后的区域扩张表现出两种不同的模式:克隆扩增和亚克隆多样化。值得注意的是,分子亚型分析揭示了一个侵袭性的 iAM 子集,其特征是亚克隆多样化、EMT增加以及 APOE+/CD163+巨噬细胞的空间富集。体外和离体实验进一步证明 APOE+ CD163+巨噬细胞通过 IGF1-IGF1R 相互作用促进肿瘤 EMT。附件受累可以预测具有更高侵袭潜力的 AMis,而 APOE 和 CD163 可作为 iAM 的预后生物标志物。总而言之,本项研究结果对 AM 的早期检测和治疗具有重要意义。
+ + + + +



