

 English
English文献解读|Cancer Cell(50.3):肿瘤细胞内在的表观遗传失调塑造了癌症相关成纤维细胞的异质性,从而在代谢上支持胰腺癌
✦ +
+
论文ID
原名:Tumor cell-intrinsic epigenetic dysregulation shapes cancer-associated fibroblasts heterogeneity to metabolically support pancreatic cancer
译名:肿瘤细胞内在的表观遗传失调塑造了癌症相关成纤维细胞的异质性,从而在代谢上支持胰腺癌
期刊:Cancer Cell
影响因子:50.3
发表时间:2024.03.31
DOI号:10.1016/j.ccell.2024.03.005
背 景
胰腺导管腺癌 (PDAC) 中的肿瘤微环境 (TME) 涉及癌症相关成纤维细胞 (CAF) 的大量积累,作为宿主对肿瘤细胞反应的一部分。 PDAC 中转录多样性 CAF 群体的起源和功能仍然知之甚少。
实验设计

结 果
01

SETD2缺陷的胰腺肿瘤中线粒体氧化磷酸化 (OXPHOS)发生激活
SETD2突变在包括 PDAC 在内的各种肿瘤中普遍存在。为了进一步深入研究 SETD2 在肿瘤代谢中的作用,研究者团队使用 TCGA-PAAD 和 QCMG-PAAD 数据集进行了基因集富集分析 (GSEA)。在 PDAC 患者中,SETD2突变和KRAS突变表现出 OXPHOS 的显著富集(图 1A-B)。此外,在具有低SETD2表达水平(SET D2low 与 SETD2high)的 PDAC 中也观察到类似的结果(图 1C)。有趣的是,SETD2缺失对OXPHOS升高的影响从PDAC扩展到其他实体肿瘤,如肺腺癌(LUAD)、结直肠腺癌(COAD)、前列腺腺癌(PRAD)和肾乳头状细胞癌(KIRP),突出了SETD2在肿瘤进展过程中代谢重塑中的潜在作用。
使用基因工程小鼠模型(GEMM),他们对10周龄的具有Setd功能(Pdxcre;LSL-KrasG12D,KC)和Setd2缺陷型(Pdxcre;LSL-KrasG12D:Setd2f/f,KSC)小鼠的胰腺肿瘤进行了无偏倚的单细胞转录组分析(scRNA-seq)。使用均匀流形近似和投影(UMAP),降维得到6个细胞聚类,共有6190个细胞,包括腺泡和导管细胞、内皮细胞、成纤维细胞、巨噬细胞、中性粒细胞以及T和NK细胞(图1D)。
为了进一步解剖腺泡/导管细胞的细微差异,他们将它们重新聚集成5个亚群。其中,表达Amy2和Prss1标记的腺泡细胞亚群占主导地位,占细胞总数的一半以上。此外,一个定义为腺泡导管化生(ADM)的亚群,共同表达腺泡和导管标记物(Sox9、Krt19和Epcam)(图1E-F)。三个亚聚类仅表示导管标记,分别表示为导管#1、#2和#3(图1E-F)。基于分区的图抽象显示了从腺泡到ADM的主要伪时间轨迹,分化为不同的导管细胞亚群,特别是导管#2和#3(图1G)。值得注意的是,导管#3主要来自KSC肿瘤(图1H)。GO分析显示,每个导管亚聚类的特征基因都有明显的富集,炎症反应、脂质代谢过程、OXPHOS和细胞分化在导管#3中尤为显著(图1I)。SCENIC(单细胞调控网络推断和聚类)分析强化了这些发现,表明干细胞相关转录因子(TF)(例如Klf4、Klf5和Gata5)、上皮-间质转化(EMT)相关TF Twist1和脂质代谢调节因子Pparg在导管#3中富集(图1J)。与此一致,GSEA分析证实了KSC与KC在导管恶性细胞(导管#1、#2和#3的组合)中OXPHOS和脂肪酸代谢的富集(图1K)。OXPHOS作为线粒体代谢的核心过程,通过对KSC和KC肿瘤的恶性肿瘤细胞进行分类,进一步验证了其代谢活性。与scRNA-seq结果一致,ksc来源的肿瘤细胞表现出增强的线粒体呼吸,这表明氧气消耗率(OCR)增加(图1L)。
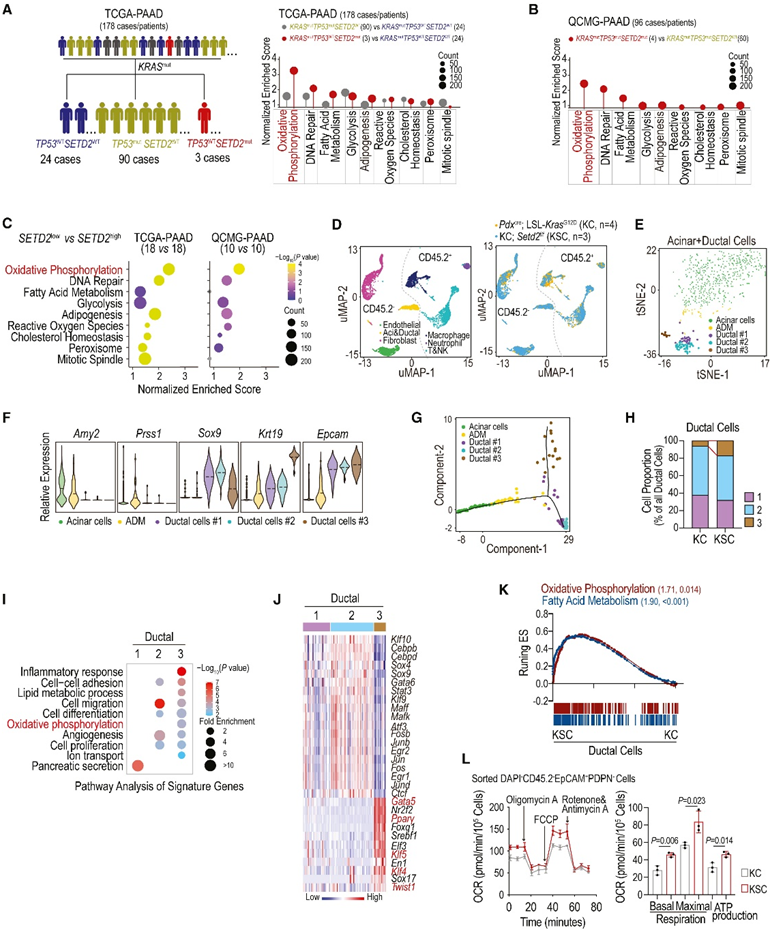
图1. 线粒体 OXPHOS 在人和小鼠SETD2缺陷的胰腺肿瘤中发生激活。
(A-B) 基于指示的突变状态对 TCGA-PAAD 和 QCMG-PAAD 数据集中的基因表达进行基因集富集分析 (GSEA)。(C) TCGA-PAAD和qcg - paad数据集中SETD2low和SETD2high组的GSEA比较。(D) UMAP可视化。(E) 腺泡和导管细胞的重新聚集,以 t-SNE 图表示。(F) 小提琴图显示所选标记基因的标准化表达。(G) 腺泡和导管亚群的伪时间轨迹分析。(H) KC 和 KSC 中导管亚聚类的比例。(I) 对指定导管亚聚类的基因进行 GO 富集分析。(J) 热图通过 SCENIC 分析显示每个导管亚聚类中富集的转录因子。(K) KC 和 KSC 之间所有导管细胞中基因表达的 GSEA 分析。(L) 从 KSC 和 KC 小鼠胰腺组织中分选的肿瘤细胞的耗氧率 (OCR)。
02
增强线粒体OXPHOS驱动setd2缺陷肿瘤的进展
为了评估OXPHOS升高对setd2缺陷胰腺肿瘤的影响,他们进一步研究Gboxin(一种选择性OXPHOS抑制剂,靶向具有高线粒体膜电位的肿瘤细胞)。在胰腺肿瘤衍生的类器官中,与KC肿瘤相比,来自KSC肿瘤的Gboxin表现出更显著的大小减少(图2A)。增强的OXPHOS依赖性通常是肿瘤干性的特征。来自KSC肿瘤的类器官显示出升高的TMRM水平和干细胞相关的tf(例如Klf4和Klf5)(图2B-C)。在这里,Gboxin处理导致KSC类器官的线粒体膜电位和干性显著降低(图2B-C)。他们使用S-Gboxin(一种为体内治疗设计的改良形式的Gboxin),对肿瘤生长和肝脏转移具有显著抑制作用,并显著延长了KSC小鼠的总体生存期(图2D-G)。S-Gboxin对肿瘤细胞中OXPHOS活性有明显的抑制作用(图2H)。在原位胰腺肿瘤模型中,S-Gboxin显著抑制了Setd2KO肿瘤的生长,而对Setd2WT肿瘤的影响最小(图2I)。此外,S-Gboxin治疗显著延长了原位Setd2KO肿瘤小鼠的总生存期,而在Setd2WT组中效果有限(图2J)。与这些观察结果一致,S-Gboxin在Setd2KO PDAC细胞中显著损害OXPHOS活性,而对Setd2WT细胞的影响很小(图2K)。这些结果强调了OXPHOS升高在驱动Setd2缺陷胰腺肿瘤进展中的关键作用。
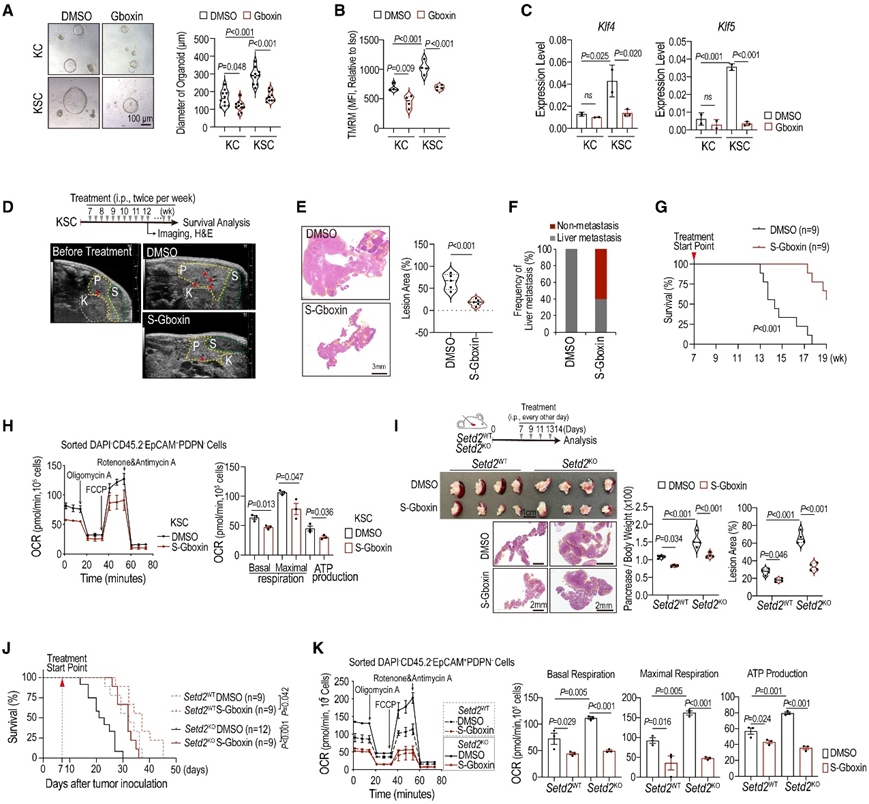
图2. Setd2缺陷肿瘤进展中线粒体 OXPHOS 增强。
(A) 用 DMSO 或 Gboxin 处理 48 小时的 KC 和 KSC 类器官的代表性图像和定量分析。(B) 所示类器官细胞的 TMRM(四甲基罗丹明甲酯)水平。(C) qPCR 测定指定类器官中Klf4和Klf5 mRNA 水平。(D) KSC 小鼠在 DMSO 或 S-Gboxin 治疗前和治疗期间的 B 扫描超声图像。(E)胰腺组织的 H&E 染色和定量。(F) KSC小鼠的肝转移统计。(G) 用 DMSO 或 S-Gboxin 治疗的 KSC 小鼠的总体存活率。(H) 从 KSC 胰腺组织中分选的肿瘤细胞的 OCR。(I) H&E 染色。(J) 用 DMSO 或 S-Gboxin 治疗的原位肿瘤小鼠的总生存率。(K)肿瘤细胞的 OCR。
03
富含脂质的 TME 在Setd2缺陷的胰腺肿瘤中支持线粒体FAO-OXPHOS
葡萄糖、谷氨酰胺和脂肪酸都是线粒体的呼吸底物。为了揭示Setd2KO PDAC细胞中OXPHOS升高的主要原因,他们从原位肿瘤中对Setd2WT和Setd2KO细胞进行了新分类,并使用选择性代谢抑制剂进行处理:UK-5099(线粒体丙酮酸载体,MPC抑制剂),BPTES(谷氨酰胺酶抑制剂),依托莫西(肉碱棕榈酰转移酶1A, CPT1α抑制剂),以及两者的组合(图3A)。值得注意的是,setd2缺陷的PDAC细胞的基础OCR呼吸由依托莫西显著抑制,这突出了脂肪酸β-氧化(FAO)来维持OXPHOS的偏好(图3A)。在体内,所有抑制剂都抑制了Setd2KO肿瘤的生长,其中依托莫西的抑制作用最强(图3B)。这些数据表明,体内缺乏Setd2的PDAC细胞表现出对氧化脂肪酸的偏好,以维持OXPHOS。
先前的研究表明,富含脂质的器官环境(例如乳腺和大脑)中的肿瘤细胞更喜欢吸收脂质以进行增殖和转移。通常情况下,正常胰腺和胰腺肿瘤都是富含脂质的组织,但他们意外地发现原位Setd2KO肿瘤中存在大量脂肪细胞样细胞(图 3 C),其标志是脂滴(ld)上的periilipin 1 (PLIN1)和periilipin 2 (PLIN2)水平升高(图 3 D)。免疫荧光染色发现,这些ld阳性细胞主要与泛caF标记物podoplanin (PDPN)共染色,而与CK19(胰腺肿瘤细胞的标记物)共染色较少,表明这些ld阳性细胞主要是caF(图3D)。PLIN1信号仅在脂肪细胞样的PDPN+细胞中观察到,而PLIN2信号也存在于PDPN+细胞中,但没有脂肪细胞样的形态学改变(图3D)。与来自Setd2WT肿瘤的PDPN+细胞相比,来自Setd2KO肿瘤的PDPN+细胞含有升高的中性脂(由BODIPY指示)、细胞内甘油三酯(tg)、非酯化脂肪酸(NEFA)和胆固醇(图3E-F)。
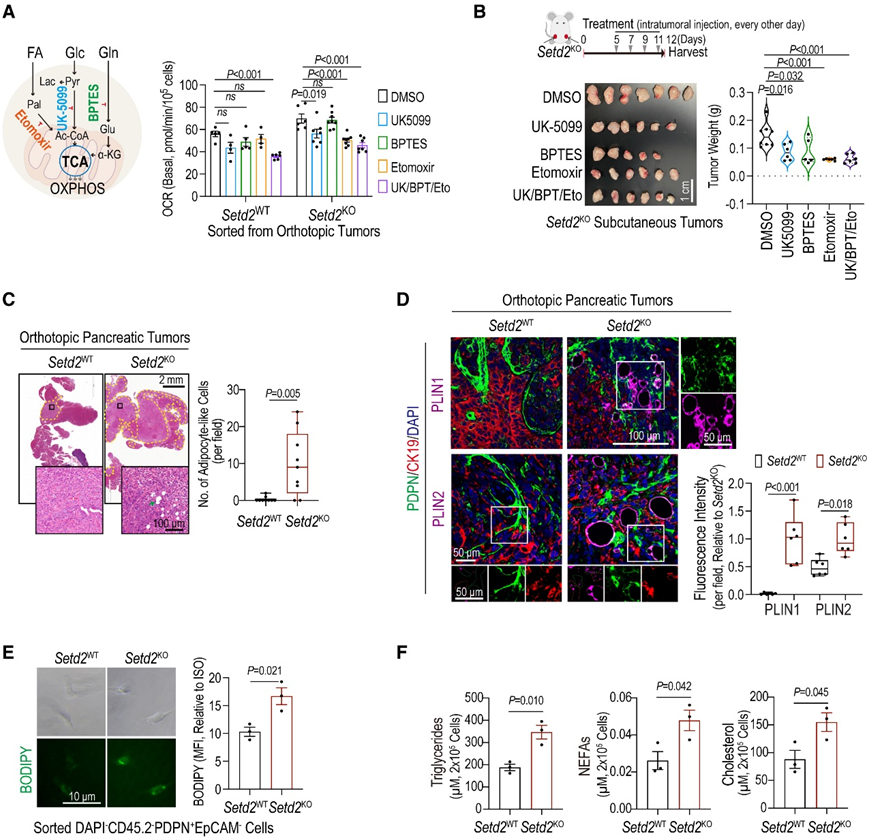
图3. 富含脂质的 TME 在Setd2缺陷的胰腺肿瘤中支持线粒体FAO-OXPHOS。
(A) DMSO、UK5099、BPTES、Etomoxir及其组合处理下Setd2 WT和Setd2 KO细胞的示意图和基础线粒体呼吸水平。(B)用 UK5099、BPTES、Etomoxir、组合或 DMSO 治疗的皮下Setd2 KO肿瘤。(C) H&E 染色。(D)免疫荧光染色和统计分析。(E) BODIPY 493/503 水平的代表性图像和统计分析。(F) 在来自原位Setd2 WT和Setd2 KO胰腺肿瘤的CAF 中测量的细胞甘油三酯 (TG)、非酯化脂肪酸 (NEFA) 和胆固醇。
04
scRNA-seq 分析识别了Setd2缺陷型胰腺肿瘤中具有脂质负载特征的独特 CAF 子集
为了探索富含脂质的基质增加的起源,他们重新分析了来自 GEMM 的 scRNA-seq 数据,识别出具有独特特征的 4 个 CAF 亚聚类 (C0-C3)(图 4 A)。聚类(C)3 CAF在KC肿瘤中普遍存在,而C2在KSC肿瘤中占主导地位(图4B)。伪时间排序揭示了C3和C2不同的进化轨迹(图4C),表明分化、表型和功能的多样性。根据程序间相关评分,C3 caf表现出KPC (pdxcre;LSL-KrasG12D;TP53R172H/+)小鼠,Mfap4、Col1a1和Col1a2的表达较高,而C1亚群表现出类似于iCAF的转录组学特征(图4D)。C2中最富集的标记基因为Abca8a、Cxcl14和Lpl(图4C-D)。GSEA分析显示C3特征基因在氧化磷酸化、缺氧和胶原纤维组织途径中富集,进一步证实了它们与myCAF的相似性(图4E)。相比之下,C2特征基因显示出与ABC转运蛋白和脂肪形成相关的独特转录组,表现出脂质代谢特征(图4E)。
接下来,他们验证了setd2缺失的KSC和setd2精通的KC和KPC小鼠肿瘤中CAF的异质性。scRNA-seq结果显示,Acta2(编码α-SMA)在C3 caF中特异性表达,而Fap和Abca8a主要存在于C2 caF中(图4F)。值得注意的是,α-SMA+ CAF在KSC肿瘤中很少见,而在KC和KPC肿瘤中则很常见(图4G-H)。FAP+ CAF在KSC肿瘤中显著增加,位于肿瘤细胞近端,而PDPN+FAP−α-SMA− CAF位于KC、KPC和KSC肿瘤细胞远端(图4H)。总的来说,具有脂质代谢转录组学特征的C2 CAFs主要位于KSC肿瘤中,其独特的空间分布表明它们可能与肿瘤细胞有密切的相互作用。
他们分别使用不含死细胞、上皮细胞和免疫细胞的DAPI−CD45.2 −EpCAM−细胞组分。ABCA8a作为C2 caf的关键标记,与FAP一起对C2 caF进行分类(图4C)。C2 CAF定义为PDPN+ABCA8a+FAP+细胞,PDPN+ABCA8a−FAP−细胞作为对照细胞。如图所示,ABCA8a+FAP+ CAF的比例在KSC肿瘤中明显较高(图4I)。更重要的是,与ABCA8a−FAP− CAF相比,从KSC肿瘤中分选出来的ABCA8a+FAP+ CAF含有更多的中性脂质,以及更丰富的细胞内tg和nefa,证实了它们的富脂表型(图4J-K)。
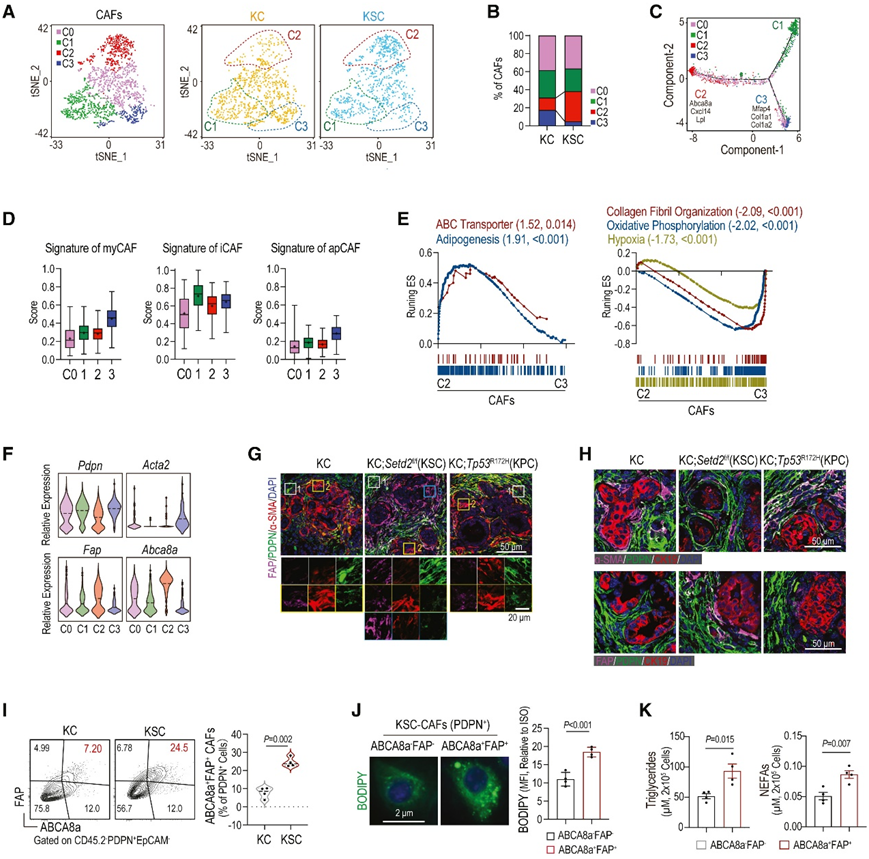
图4. scRNA-seq 分析在Setd2缺陷的胰腺肿瘤中鉴定出具有富含脂质特征的独特 CAF 子集。
(A) t-SNE 图中显示的 CAF 重新聚类,其中圈出 C1、C2 和 C3 CAF 亚聚类。(B) KC 和 KSC 中 CAF 亚聚类的相对细胞比例。(C) 列出了 C2 和 C3 CAF 的前 3 个特征基因的 CAF 的伪时间轨迹分析。(D) GSEA 注释程序与之前报道的 CAF 特征的相似性。(E) C2 和 C3 CAF 之间基因表达的 GSEA。(F) 小提琴图显示不同 CAF 亚聚类中Pdpn、Acta2、Fap和Abca8a的标准化表达。(G-H) KC、KSC 和 KPC 胰腺的免疫荧光染色。(I)流式细胞分析和统计分析。(J) BODIPY 493/503 的代表性图像和流式细胞分析。(K) 在来自 KSC 的 ABCA8a + FAP +和 ABCA8a - FAP - CAF 中测量的细胞 TG 和 NEFA。
05
肿瘤细胞衍生的 BMP2 驱动脂质负载的 CAF 分化
CellChat和Cellphone (CellphoneDB)研究了setd2缺陷胰腺肿瘤中富含脂质的CAF,结果显示BMP信号是关键因素(图5A)。BMP2在导管3亚聚类中主要表达,并与C2 caf中的受体表现出强烈的相互作用,表明BMP2信号从导管细胞传递到caf(图5B-C)。此外,C2 caF表现出BMP4水平升高,并表现出显著的BMP4配体与受体相互作用,表明在setd2缺陷胰腺肿瘤的cas中存在自分泌BMP4信号传导(图5B-C)。
已知BMP2和BMP4诱导肌成纤维细胞向脂质储存细胞的反式分化。在条件培养基(CM)中检测来自Setd2KO肿瘤细胞的主要CAF祖细胞,胰腺星状细胞(PSC)和骨髓间充质干细胞(BMSC),发现脂质负荷表型和脂质相关基因表达上调(图5D-E)。用中和抗体阻断BMP2有效地阻止了暴露于Setd2KO CM的psc中的脂质积累(图5F-G)。
为了进一步研究BMP2/BMPR轴在体内脂质负载ABCA8a+FAP+ CAF形成中的作用,他们分别在Setd2WT和Setd2KO PDAC细胞中沉默BMP2。Setd2KO-shBmp2 PDAC细胞的CM显著降低了PSC的脂质储存,而Setd2WT-shBmp2组无显著影响(图5H)。重要的是,在原位肿瘤模型中,Bmp2沉默显著阻碍了Setd2KO肿瘤的进展,并伴随着ABCA8a+FAP+ CAF形成的减少(图5I-J)。
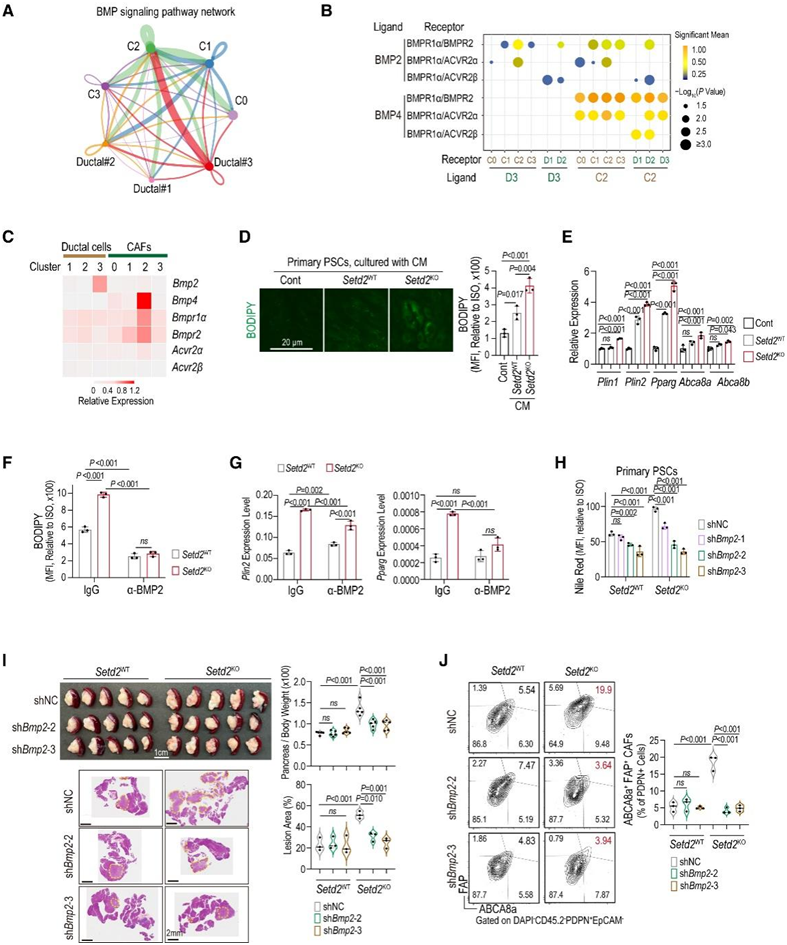
图5. 肿瘤细胞衍生的 BMP2 驱动脂质负载的 CAF 分化。
(A) 通过 CellChart 定量推断成纤维细胞和导管细胞之间 BMP 信号网络的相互作用。(B) 使用 CellphoneDB 绘制导管细胞和成纤维细胞之间显著受体-配体相互作用的点图。(C) 点图显示 scRNA-seq 中导管和 CAF 亚群中所示基因的相对表达水平。(D) 用Setd2 WT和Setd2 KO胰腺肿瘤细胞的条件培养基 (CM) 和对照培养基培养的 PSC 中 BODIPY 493/503 水平的代表性图像和统计分析。(E) 对 (D) 的 PSC 中指定基因进行 qPCR 测定。(F) PSC 中的 BODIPY 493/503 水平。(G) 对 (F) 的 PSC 中指定基因进行 qPCR 测定。(H) 具有来自Setd2 WT和Setd2 KO胰腺肿瘤细胞(w/或 w/o Bmp2敲低)的 CM 的 PSC 中的尼罗红水平。(I)胰腺重量、H&E 染色和病变面积百分比。(J) 来自指定胰腺肿瘤的 ABCA8a + FAP + CAF的细胞比例的代表性图和统计分析。
06
脂质负载的CAF通过ABCA8a转运脂质维持肿瘤生长
最近的研究揭示了CAF在提供氨基酸、核苷和外泌体来滋养肿瘤细胞中的作用。在空间上,他们观察到脂质负载的CAF与KSC中的恶性细胞之间存在密切的相互作用(图4G-H),这促使他们研究脂质负载的CAF是否将脂质转运到肿瘤细胞中以支持其OXPHOS和生长。通过离体脂质脉冲追踪实验,他们证明了与来自setd2 -正常肿瘤的CAF相比,从setd2缺陷肿瘤中分离的CAF有效地将更多的荧光长链脂肪酸(LCFA和 BODIPY-C16)转移到肿瘤细胞中(图6A-C)。
为了了解脂质转移的机制,他们重点研究了ABCA8a,它在脂质负载的CAF中升高,在脂质、胆固醇和牛磺酸(TCA)的外排中起作用。永生化PSC(iPSC)已广泛用作研究胰腺caf作用的模型。通过操纵iPSC中Abca8a的表达,他们发现Abca8a的过表达促进了BODIPY-C16从iPSC外排并转运到PDAC细胞中(图6D-E)。相反,Abca8a的缺失减少了BODIPY-C16的外排及其向PDAC细胞的转运(图6D-E)。
先前的研究表明,富含脂质的细胞通过游离脂肪酸或细胞外囊泡将其脂质转移到周围细胞。他们将BODIPY-C16标记的iPSC中的CM根据大小分为三部分:游离脂肪酸(<10 kDa)和脂肪酸携带载体:中型(10 -100 kDa)和大型(>100 kDa)馏分。值得注意的是,在大剂量(>100 kDa)和中等剂量(10-100 kDa)处理的PDAC细胞中,荧光信号增加(图6F-G)。此外,iPSC中ABCA8a的过表达增加了bodipyc16向PDAC细胞的转运,特别是在中等大小的CM部分,类似于ABC家族介导的高密度脂蛋白(HDL)颗粒的大小(图6F-G)。他们研究了caf中ABCA8a转运体对体内肿瘤生长的影响,发现在Setd2WT和Setd2KO组中,iPSC与PDAC细胞共接种均显著促进了肿瘤生长(图6H)。在Setd2KO肿瘤中,iPSC中Abca8a的缺失完全消除了它们的致瘤功能,而共接种Abca8a过表达的iPSC则加速了Setd2KO肿瘤的生长(图6H)。植入Abca8a−/−小鼠的setd2缺陷PDAC细胞比野生型小鼠生长更慢(图6I-K)。
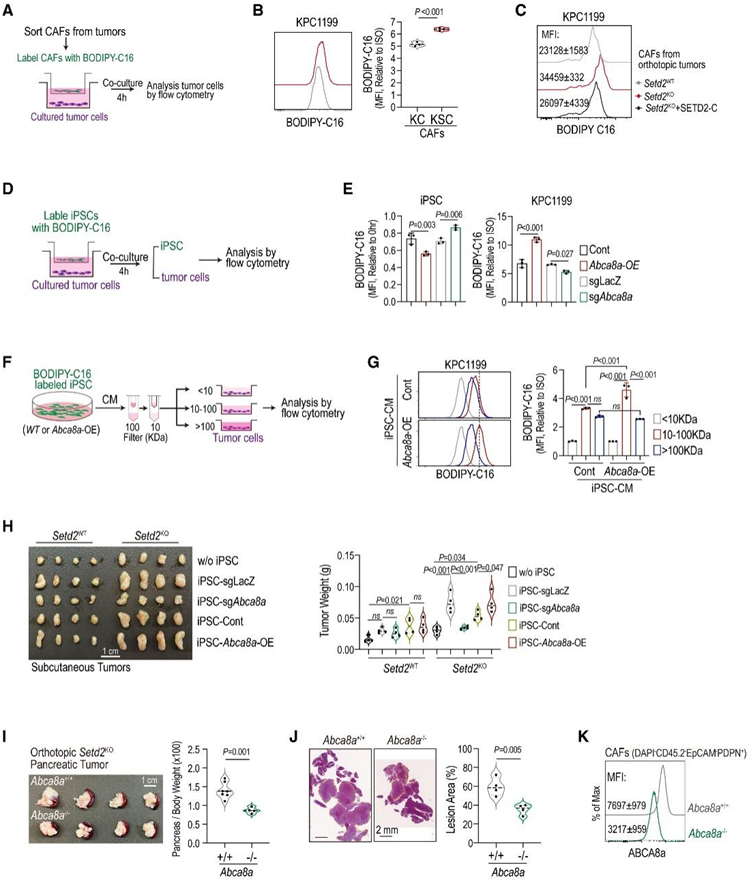
图6. 富含脂质的 CAF 通过 ABCA8a 运输脂质以维持肿瘤生长。
(A) BODIPY-C16从CAF转移到肿瘤细胞的示意图。(B-C) KPC1199 与来自指定肿瘤的 BODIPY-C16 标记的 CAF 共培养中的 BODIPY-C16 水平。(D-E) Abca8a过表达 (Abca8a -OE) 和Abca8a耗尽 (sg Abca8a ) iPSC 与 KPC1199 共培养的示意图,评估 BODIPY-C16 水平。(F-G)用 BODIPY-C16 标记的 Abca8a -OE iPSC 的示意图,以及用于评估 BODIPY-C16 向肿瘤细胞转运的条件培养基 (CM) 级分。(H) 肿瘤重量。(I-J)对相对胰腺重量和病变面积进行定量。(K) 来自所示小鼠的原代 CAF 上的 ABCA8a 水平。
07
Setd2丢失引起的异位 H3K27Ac 沉积导致胰腺肿瘤转录组重编程
他们观察到KSC 小鼠的Setd2KO原位肿瘤(图 7 A)和胰腺肿瘤病变中 H3K27me3 和 H3K27Ac 水平显著增加,同时 H3K36me3 水平降低(图 7B)。巧合的是,Setd2 KO肿瘤表现出乙酰辅酶 A 水平的特异性增加,乙酰辅酶A 是线粒体代谢的关键代谢物(图 7 C)。他们进一步对从原位肿瘤分选的Setd2WT和Setd2KO PDAC 细胞中的 H3K27Ac 和 H3K27me3 进行了 CUT&Tag 分析(图7D)。RNA-seq 数据与 H3K27Ac 和 H3K27me3 峰相关基因集的交叉显示,基因体上具有 H3K27Ac 增益的基因表现出显著的整体上调,而与 H3K27me3 的关联较少(图 7 E)。重点关注339个基因体和启动子区域H3K27Ac增加的重叠基因,它们与ecm受体相互作用、谷胱甘肽代谢、局灶黏附、tgf-β信号传导、细胞迁移和OXPHOS有关(图7F)。值得注意的基因包括Bmp2、空泡内吞调节基因(Cav2、Cavin2和Cavin3)、谷胱甘肽代谢基因(Idh1、Gpx2、Gsto1、Gsto2、Mgst3和Nqo2)、fao -oxphos相关基因(Acadl和Acsl5)和干细胞相关tf(图7F-H)。
他们评估了DEGs上的h3k27ac增益是否直接依赖于H3K36me3信号的丢失。在Setd2WT PDAC细胞中,只有约10%的deg(339个deg中的33个,如Bmp2、Idh1和Klf4)在基因体上有H3K36me3沉积,表明setd2介导的H3K36me3缺失在整体上独立地改变了H3K27Ac(图7H)。整合基因组观察器(IGV)显示,Setd2缺失后启动子和基因体(前两个内含子和/或外显子区域)的H3K27Ac峰值增加,与AP-1家族tf、JunB和Fosl1结合一致(图7H)。他们进一步研究了Setd2KO PDAC中H3K27Ac增益对进展的影响。在原位肿瘤中,JQ1部分抑制了Setd2KO肿瘤生长,抑制了肿瘤细胞中富含脂质的TME和OXPHOS活性,对Setd2WT组的影响最小(图7I-K)。JQ1还抑制了Setd2KO中异位H3K27Ac沉积上调的基因表达(图7L)。
综上所述,这些发现表明Setd2缺失诱导异位H3K27Ac沉积,导致转录组重编程,改变肿瘤进展的代谢适应。
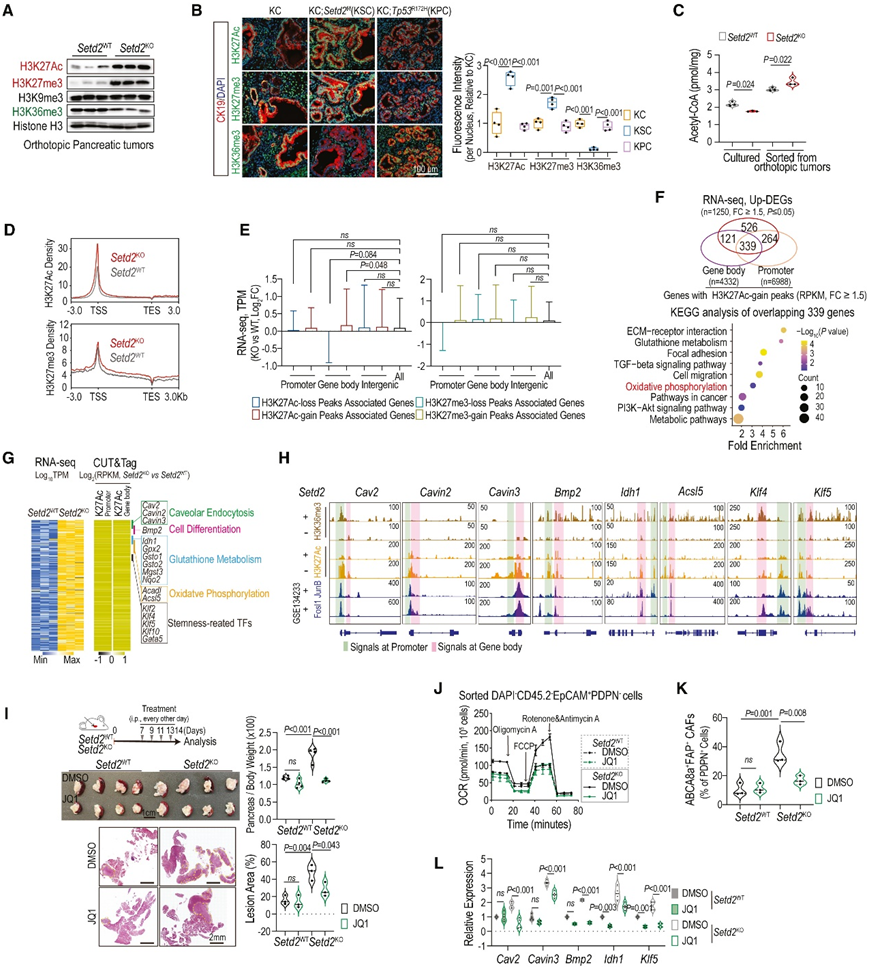
图7. Setd2丢失引起的异位 H3K27Ac 沉积导致胰腺肿瘤转录组重编程。
(A) 原位Setd2 WT和Setd2 KO胰腺肿瘤中的组蛋白修饰标记。(B) KC、KSC 和 KPC 小鼠胰腺切片中 H3K27Ac、H3K27me3 或 H3K36me3(绿色)和 CK19(红色)的免疫荧光染色。(C) 来自原位肿瘤的所示培养或分选的 PDAC 细胞中的细胞乙酰辅酶 A 水平。(D) 从转录起始位点 (TSS) 上游 3000 bp 到转录终止位点 (TES) 下游的 H3K27Ac 或 H3K27me3 的密度。(E) 箱线图显示具有 H3K27Ac 或 H3K27me3 差异沉积的指定基因集的归一化转录变化。(F) 与显示 H3K27Ac 增益峰的基因重叠的上调基因的维恩图,并对重叠基因进行 KEGG 通路分析。(G) 热图显示 339 个重叠基因的启动子和基因体区域中 H3K27Ac 沉积的 mRNA 水平和差异倍数。(H) Setd2 WT和Setd2 KO肿瘤细胞中 H3K27Ac 和 H3k27me3 CUT&Tag 信号的 IGV 屏幕截图,以及来自胰腺肿瘤细胞的 JunB 和 Fosl1 ChIP 信号。(I)定量胰腺重量和病变面积。(J)胰腺原位肿瘤中分选的肿瘤细胞的 OCR。(K)通过流式细胞对来自所示肿瘤的ABCA8a + FAP + CAF的细胞比例进行统计分析。(L)使用 DMSO 或 JQ1 治疗的Setd2 WT和Setd2 KO肿瘤中指定基因的相对 mRNA 水平。
08
SETD2/H3K36me3 水平、CAF 亚型和 OXPHOS 依赖性在人 PDAC 中的临床意义
为了加强临床相关性,他们研究了人 PDAC (hPDAC) 中SETD2/H3K36me3 水平与 ABCA8+ FAP+ CAF的相关性。与动物模型一致,H3K36me3 与hPDAC 中的 ABCA8+ FAP+ CAF 呈负相关,经流式细胞分析验证(图8A-C)。此外,他们观察到H3K36me3低hPDAC组织中存在脂肪细胞样细胞和分化程度较低的肿瘤细胞(图8D)。此外,免疫组化染色显示H3K36me3与FAP水平呈负相关,而H3K36me3与PDPN水平呈正相关(图8E),表明ABCA8+FAP+ CAF的存在,而KSC小鼠胰腺组织中的CAF(PDPN+细胞)总量低于KC小鼠。
SETD2突变或低mRNA水平的PDAC表现出更高的OXPHOS活性。使用患者来源的异种移植(PDX)模型,他们发现,与hPDAC一致,H3K36me3与ABCA8+FAP+ CAF负相关,并且在H3K36me3-low的PDAC细胞中显示出更高的线粒体电位。随后,他们给H3K36me3low和H3K36me3high胰腺pdx给予S-Gboxin和JQ1处理(图8F)。正如预期的那样,H3K36me3水平较低的PDAC对S-Gboxin和JQ1表现出更高的敏感性(图8F)。相反,H3K36me3水平较高的PDAC对S-Gboxin反应温和,对JQ1无显著反应。
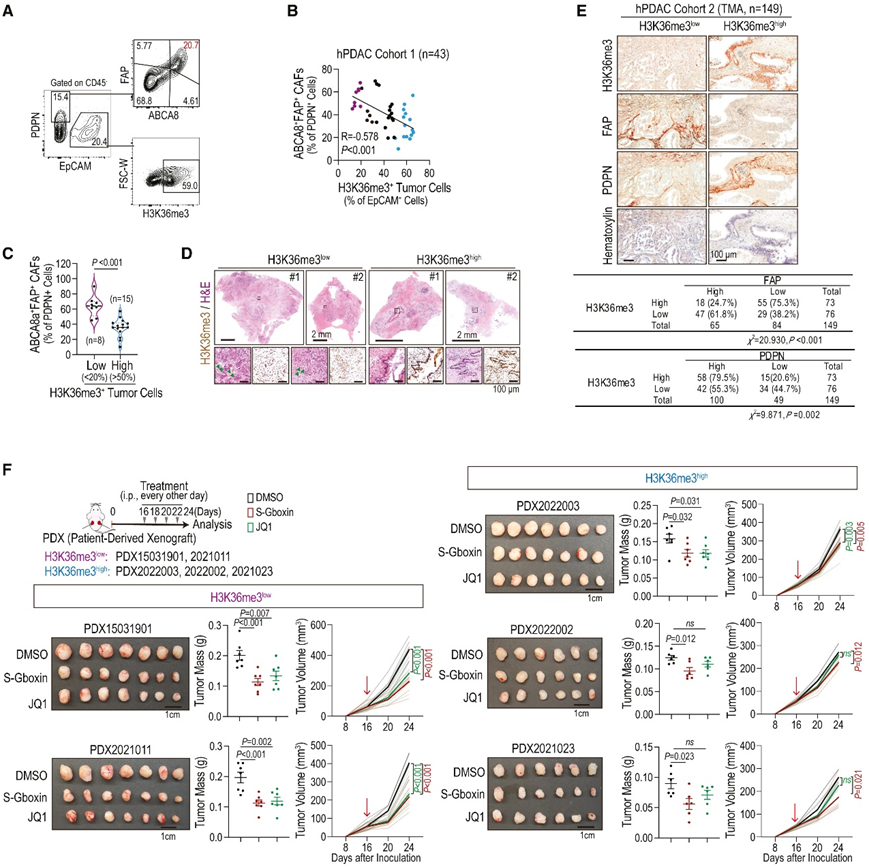
图 8 .人 PDAC 中 SETD2/H3K36me3 水平、CAF 亚型和 OXPHOS 敏感性的临床意义。
(A)对 PDAC 患者原发肿瘤组织中ABCA8a + FAP + CAF 和 H3K36me3 +肿瘤细胞的细胞比例进行流式细胞分析。(B) ABCA8a + FAP + CAF 与 H3K36me3 +肿瘤细胞的细胞比例之间的相关性。(C) H3K36me3 +肿瘤细胞水平低和高的 PDAC 患者中ABCA8a + FAP + CAF 的细胞比例。(D) PDAC 患者原发性肿瘤组织中 H3K36me3 的代表性 H&E 和免疫组织化学染色。(E) PDAC TMA 中 H3K36me3、FAP 和 PDPN 多重免疫组织化学染色的代表性图像。(F) 在患者来源的异种移植 (PDX) 模型中进行治疗,评估肿瘤质量和体积。
+ + + + + + + + + + +
结 论
通过scRNA-seq,研究者团队在Setd2缺陷的胰腺肿瘤中鉴定出由 ABCA8a 标记的富含脂质的 CAF 亚群。研究结果表明,肿瘤固有的SETD2缺失通过 H3K27Ac 的异位增益释放 BMP2 信号传导,导致 CAF 向富含脂质的表型分化。然后,充满脂质的 CAF 通过 ABCA8a 转运蛋白为线粒体氧化磷酸化提供脂质,从而促进肿瘤进展。总之,这项研究将 CAF 异质性与肿瘤细胞的表观遗传失调联系起来,强调了 CAF 和胰腺肿瘤细胞之间以前未认识到的代谢相互作用。
+ + + + +



