

 English
English文献解读|Cancer Cell(50.3):蛋白质组学分析表明子宫内膜癌的药物途径
✦ +
+
论文ID
原名:Proteogenomic insights suggest druggable pathways in endometrial carcinoma
译名:蛋白质组学分析表明子宫内膜癌的药物途径
期刊:Cancer Cell
影响因子:50.3
发表时间:2023.09.11
DOI号:10.1016/j.ccell.2023.07.007
背 景
子宫内膜癌(EC)是发达国家最常见的妇科恶性肿瘤,EC结果恶化是由于侵袭性 EC 组织型的发生率增加,特别是在黑人和西班牙裔女性中。对于高危 EC 患者,辅助放疗和细胞毒性化疗可以降低疾病复发的可能性并提高生存率。但放疗或细胞毒性化疗能否改善处于疾病进展或复发中等风险的 EC 患者的预后仍不太清楚。
实验设计
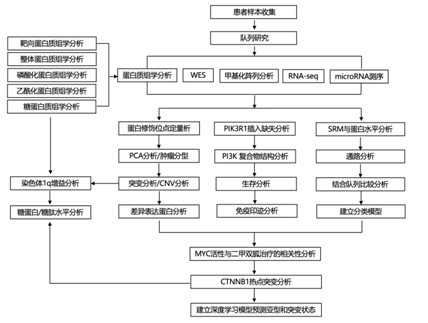
结 果
01

独立 EC 队列的蛋白质组景观
研究者团队使用包括全基因组测序在内的 10 个组学平台,对 138 个 EC 肿瘤(包括 119 个子宫内膜样肿瘤、13 个浆液性肿瘤和 3 个透明细胞肿瘤)、匹配的正常血液样本和来自健康捐献者的 20 个富集正常子宫内膜样本进行了蛋白质组学分析、全外显子组测序(WES)、甲基化阵列、整体RNA测序(RNA-seq)、microRNA测序、靶向蛋白质组学分析、全蛋白质组学分析、磷酸化蛋白质组学分析、乙酰基化蛋白质组学分析和糖蛋白质组学分析(图S1A-SL),总共对 10135 个蛋白质和 25300 个磷酸化位点、5556 个乙酰化位点和 6513 个糖基化位点进行了定量(图 1 A)。主成分分析 (PCA) 根据全蛋白质组、磷酸蛋白质组或糖蛋白质组数据(而不是乙酰蛋白质组数据)分离肿瘤和正常样本,并且在体外肽标记技术 (TMT) 中未观察到批次效应(图 S1 E-S1H)。在探索性队列和独立队列之间观察到可定量的整体蛋白质组学和磷酸化蛋白质组学特征显著重叠,并量化了该队列中更多的乙酰化位点(图1B)。该队列中突变最多的基因包括PTEN、ARID1A、PIK3CA、PIK3R1和CTNNB1,其突变频率与探索性队列相似(图1C)。由于浆液性肿瘤的比例较高,TCGA EC队列中的突变频率较低,突变频率在三个队列中高度相关(图S1J)。他们将肿瘤分为4个基因组亚型:6个POLE、47个MSI-H、66个CNV- l和16个CNV-H肿瘤(图1C、图S1K)。基于拷贝数变异(CNV)分析发现了显著的位点扩增和缺失。几个已知的肿瘤驱动因子位于扩增区域内(如SETDB1、ECT2、ATAD2、GRB7和CCNE1)(图1D)。他们根据它们与mRNA和蛋白水平的相关性对CNV驱动基因进行优先排序(图1E),并通过比较肿瘤和富集的正常组织中的蛋白水平来筛选它们,分别从独立队列和探索性队列中确定了351和237个潜在的CNV驱动基因,其中有88个基因在它们之间显著重叠(图1E),这些基因显著富集于增殖和细胞周期相关通路(图1F)。当将肿瘤与富集的正常组织进行比较时,他们鉴定出了 1292 个上调蛋白和 1488 个下调蛋白,显著差异表达的蛋白质在独立队列和探索队列之间高度重叠(图1G)。有趣的是,两种癌症/睾丸抗原PBK和KIF2C在两个队列中均显著上调。体细胞驱动突变对蛋白质组的顺式和反式效应在不同队列中是相似的(图1H)。例如,CTNNB1突变与较高的 CTNNB1 和 LEF1 蛋白水平呈显著正相关。
在独立队列中,MLH1和HOX家族基因受到 DNA 甲基化沉默(图 S1 M)。circRNA 显示出比其宿主基因之间更强的正相关性(图 S1 N),并且 miR-200c-3p 与 circRNA 调节因子 QKI 的蛋白水平显著负相关(图 S1 O)。这些结果表明,两项EC 队列研究之间的多组学水平的许多结果高度一致。
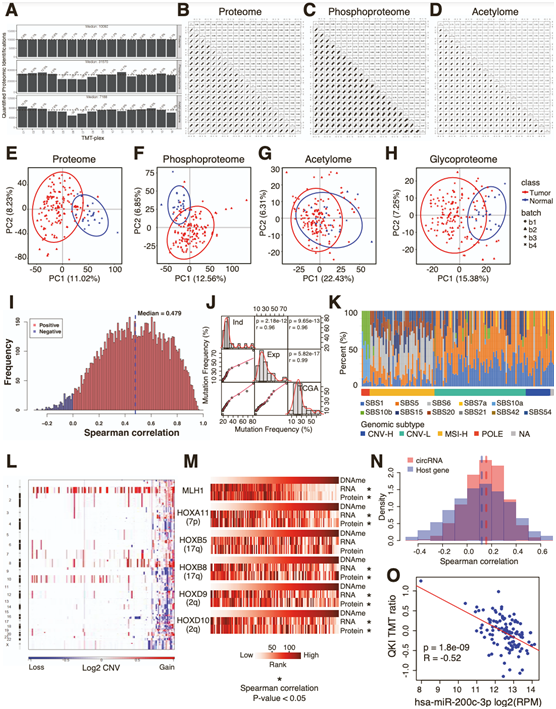
图S1. 蛋白质基因组数据。
(A)定量蛋白(上)、磷酸化位点(中)和乙酰化位点的条形图(下)。(B-D)纵向性能通过重复的蛋白质组,磷酸化蛋白质组和乙酰化蛋白质组分析在独立的TMT-11相同的患者来源的异种移植QC样本和EC研究样本的分配进行测试。(E-H)全蛋白质组,磷酸化蛋白质组,乙酰化蛋白质组和糖蛋白组的主成分分析(PCA)图,由肿瘤(红色)和正常(蓝色)样本着色。(I) mRNA与蛋白水平Spearman相关值直方图。(J)密度图显示独立队列(Ind)、探索性队列(Exp)和TCGA队列中最高突变基因突变频率的相关性。(K)独立队列的突变特征。(L)每个样本的全基因组(行)的独立队列CNV图谱(列)。(M) MLH1和HOX家族基因的DNA甲基化和mRNA/蛋白水平之间的相关性热图。(N)circRNA与宿主基因相关性的Barplot图。(O) hsa-miR-200c-3p表达circRNA调控因子QKI蛋白水平的相关性。
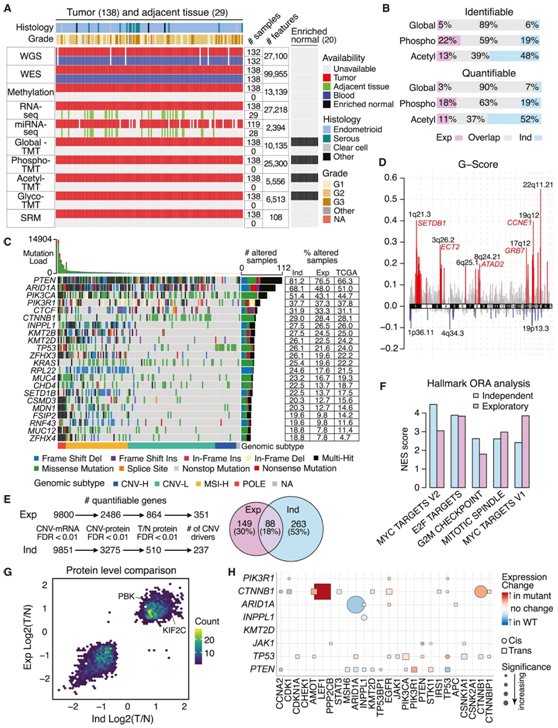
图1. 独立 EC 队列的蛋白质组图谱。
(A) 独立 EC 队列中的多组学数据可用性。(B) 探索性 (Exp) 和独立 (Ind) 队列的蛋白质组特征的鉴定和定量。(C) Oncoplot 显示通过全基因组测序确定的独立队列中最常见的突变基因。(D) 整个基因组的拷贝数变异 (CNV)(x 轴)与 G 分数(y 轴)的比较。(E) CNV驱动因素在两个CPTAC队列中的优先级。(F) 条形图显示从 CNV 驱动程序分析中富集的路径。(G) 两个 CPTAC 队列之间肿瘤/正常蛋白水平的散点图。(H) 体细胞突变(y 轴)对蛋白质表达(x 轴)的顺式/反式效应。
02
PIK3R1的插入缺失是AKT抑制反应的潜在标志物
PI3K-AKT通路在子宫内膜样肿瘤中经常发生改变,其中PTEN、PIK3CA和PIK3R1是该通路中最常见的突变基因。在约80%的子宫内膜样肿瘤中,PTEN有突变,并且大多数与PIK3CA和PIK3R1突变共发生,这两种突变是互斥的(图2A)。虽然PIK3CA在许多癌症类型中常见突变,但PIK3R1突变在EC中更常见(图1C)。有趣的是,这些识别框内插入突变在P85/iSH2结构域的两端紧密聚集,在与PIK3CA相互作用的PIK3R1区域中,它们在结构上彼此接近(图2B)。
接下来,他们研究了不同PIK3R1突变对mRNA和蛋白质水平的顺式效应(图2C-D)。识别框的插入突变与PIK3R1蛋白(PI3K的p85α亚基)水平相关(图2D),而与WT PIK3R1组相比,截断突变与较低的蛋白质水平相关。由于PIK3R1是AKT磷酸化的抑制因子,他们预计通过截断突变降低PIK3R1的水平将导致PI3K-AKT通路的激活和更差的临床结果。令人惊讶的是,在TCGA队列中,与PIK3R1截断和PIK3CA突变相比,PIK3R1框内插入突变与较差的生存期相关,尽管它们并未导致PIK3R1蛋白水平降低(图2E)。
他们假设带有插入突变序列的PIK3R1蛋白会破坏PIK3R1抑制AKT磷酸化的能力,从而导致磷酸化的AKT1水平升高。为了验证这一假设,他们首先研究了AKT1磷酸化水平与PTEN、PIK3CA和PIK3R1突变状态之间的关系。正如预期的那样,PTEN WT样品的AKT1-T308磷酸化水平明显低于PTEN突变样品(图2F)。此外,与具有PIK3CA突变或PIK3R1截断突变的癌症相比,具有插入突变的样本具有更高水平的AKT1-T308磷酸化(图2F)。由于在探索性队列中未发现该磷酸化位点,因此使用TCGA逆相蛋白阵列(RPPA)数据进一步证实了他们的假设,即在含有PIK3R1插入突变的癌症中AKT-T308磷酸化水平较高,而在PTEN WT癌症中磷酸化水平最低(图2G)。他们在另一个磷酸化位点AKT-S473上观察到同样的趋势(图2H)。
为了进一步确认PIK3R1框内插入缺失与 AKT-T308 和 AKT-S473 磷酸化之间的关系,他们通过 CRISPR-Cas9 在 EC 细胞系 HEC151 中建立了缺失 T576Del,AKT-T308 和 AKT-S473 的磷酸化水平均上调(图 2 I)。
他们接下来使用来自 DepMap 的 EC 细胞系的数据评估PIK3R1插入缺失是否可能是比PIK3CA突变更好的 AKT 抑制临床反应标记。正如预期的那样,具有 PIK3R1 插入缺失和突变 PTEN 的 EC 细胞系对 MK-2206(一种针对 AKT-S473 的成熟 AKT 抑制剂)显著更敏感(图2J)。总之,这些结果表明,具有突变PTEN 的PIK3R1插入缺失可能是 AKT 抑制反应的潜在生物标志物(图 2 K)。
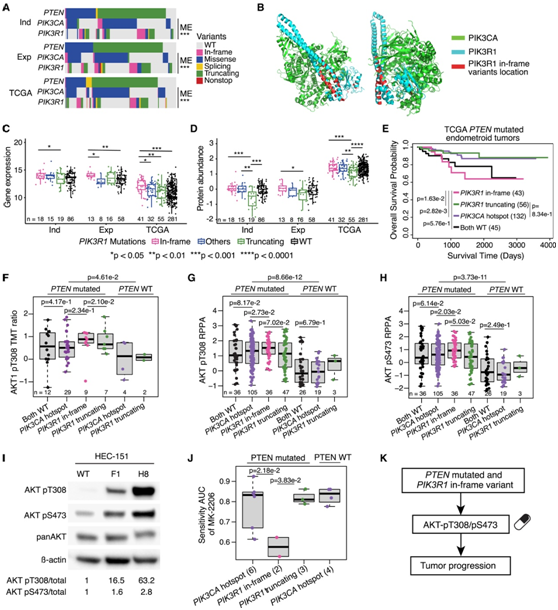
图2. PIK3R1框内插入缺失显示激活 AKT 磷酸位点的诱导。
(A)独立 (Ind)、探索性 (Exp) 和 TCGA 队列中的PTEN、PIK3CA和PIK3R1突变。(B) PI3K 复合物的 3D 结构。(C-D) 比较独立 (Ind)队列、探索性 (Exp)队列和 TCGA 队列中PIK3R1变体之间PIK3R1 mRNA和蛋白质水平的箱线图。(E)具有PIK3R1和PIK3CA变体的TCGA PTEN突变的EC患者的生存分析。(F) 比较独立队列中PIK3R1和PIK3CA变体之间 AKT1-pT308 水平的箱线图。(G-H) 比较PIK3R1和PIK3CA变体之间 AKT-pT308 (G) 和 AKT-pS473 (H) 水平的 TCGA RPPA 数据的箱线图。(I) HEC-151 细胞中 AKT pT308 和 pS473 的免疫印迹。(J) 箱线图比较 DepMap EC 细胞系对 MK-2206 的反应。(K) 显示PIK3R1突变的示意图。
03
选择性反应监测(SRM)分析准确预测抗原呈递机制(APM)状态
据报道,有效的细胞APM是免疫检查点抑制剂(ICI)反应的重要因素,不依赖于肿瘤突变负荷(TMB)。TMB-H/APM-H 肿瘤通过激活的 CD8+ T 细胞进行免疫热(immune hot)过程(图3A)。此外,JAK1变异在TMB-L肿瘤中非常罕见,这表明JAK1突变与TMB-H肿瘤有关。
目前没有临床生物标志物来确定 APM 状态。以前计算 APM 分数的方法依赖于蛋白质组范围的测量,不适合作为诊断测定。为了开发靶向蛋白质组检测,他们选择了包含 17 个 JAK-STAT 和 APM 蛋白以及 51 个其他免疫相关蛋白的组,其中每个蛋白包含两个肽。在探索性队列和独立队列中,他们观察到源自同一蛋白质的肽之间以及靶向测定和 TMT 测量之间存在很强的相关性,特别是对于水平相对较高的蛋白质(图 3B-C)。靶向测定验证了 APM 蛋白的下调,之前通过整体蛋白质组学鉴定出 TMB-H/APM-L 组中 APM 蛋白下调(图 3D-E)。通过利用这些肽测量数据,他们构建了准确预测 APM 状态的机器学习模型,仅使用两种肽即可实现 0.961 的 AUC:PSMB9−VSAGEAVVNR 和 PSMB10−LPFTALGSGQDAALAVLEDR(图3F-G)。
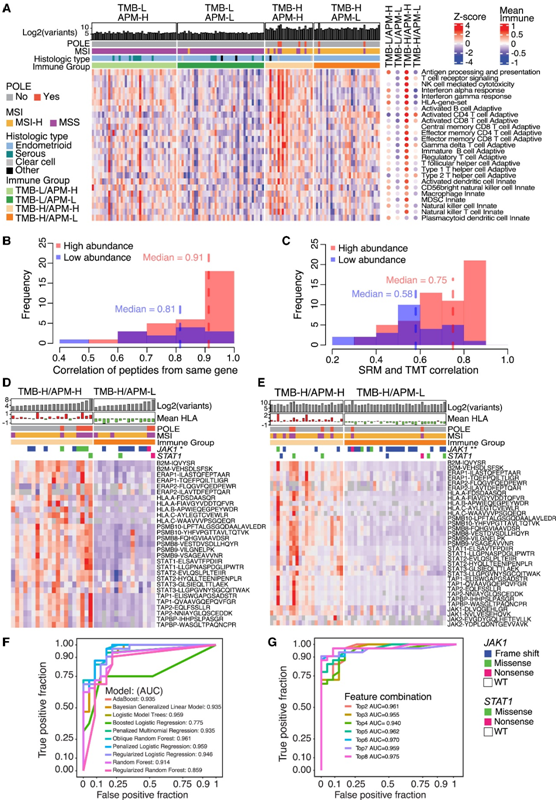
图3. 用于抗原呈递机制 (APM) 状态的选择性反应监测 (SRM) 测定。
(A) 使用蛋白质水平作为独立队列的输入,从 ssGSEA 通路评分得出的免疫亚组和相关通路的热图。(B-C) 直方图显示独立队列中来自相同基因的 SRM 肽和具有基于 TMT 的蛋白质水平的 SRM 肽之间的相关频率。(D-E) 热图显示基于 SRM 的 JAK-STAT 肽定量以及探索性和独立队列中选定的 APM 蛋白。(F) ROC 曲线显示使用两种肽PSMB10-LPFTALGSGQDAALAVLEDR 和 PSMB9-VSAGEAVVNR 的分类器的模型性能。(G) ROC 曲线显示 ORFlog 分类器在独立队列上的模型性能。
04
MYC 活性是二甲双胍治疗的目标和潜在生物标志物
肥胖和 2 型糖尿病 (T2D) 是公认的 EC 危险因素。二甲双胍治疗可以改善患有 T2D 的 EC 患者的总体生存率。然而,二甲双胍影响 EC 并可能改善患者预后的机制仍不清楚。
为了检测二甲双胍反应的分子决定因素,他们将临床表型与多组学数据相结合。基于RNA-seq数据的通路分析显示二甲双胍治疗的患者中增殖相关的MYC活性显著降低(图4A)。他们还分析了来自 DepMap 的 22 种 EC 细胞系(8 种敏感和 14 种不敏感)的反应,并发现二甲双胍敏感细胞系的MYC活性显著高于耐药细胞系(图4B)。二甲双胍处理的细胞系的几个 CMap 特征中 MYC 活性下调(图 4C),且二甲双胍敏感细胞系中的 MYC 蛋白水平较高(图4D),具有较高 MYC 活性的细胞系对二甲双胍治疗更敏感(图 4 E)。在对二甲双胍最敏感的两种细胞系中,推定的 MYC 调节转录本的减少发生得更早,程度也更大。这些结果表明二甲双胍治疗显著降低 MYC 活性,并且表达高水平 MYC 的细胞系的敏感性有增加的趋势,二甲双胍降低 EC 生长的一个重要机制是直接下调 MYC 活性。
有趣的是,高水平的 MYC 还与 EC 的 MSI-H 和 CNV-L 亚型中较差的总生存期(OS) 相关(图 4F-G)。MYC活性还与MYC免疫组织化学(IHC)评分显著相关(图4H)。因此,他们使用推断的 MYC 活性作为 MYC 蛋白水平的读数来进一步检测 MYC 和 EC 基因型/表型之间的关系。他们将患者分为 3 组:非糖尿病组、糖尿病未治疗组和糖尿病治疗组,并根据 MYC 活性从高到低对患者进行分类(图 4 I)。与 T2D 患者未经治疗的肿瘤相比,二甲双胍治疗的肿瘤的 MYC 活性水平显著降低。当他们重新评估早期的探索性队列时,观察到了类似的趋势(图 4 I)。值得注意的是,他们发现非糖尿病患者 EC 中的 MYC 活性与先前未接受二甲双胍治疗的糖尿病患者中观察到的 MYC 活性相似甚至更高(图 4 I)。他们还发现该队列中 MYC 活性与身体质量指数(BMI)呈负相关。
总的来说,这些观察结果表明,高水平的 MYC 活性可能是识别最有可能从二甲双胍治疗中受益的 EC 患者(包括非糖尿病和非肥胖患者)的生物标志物。
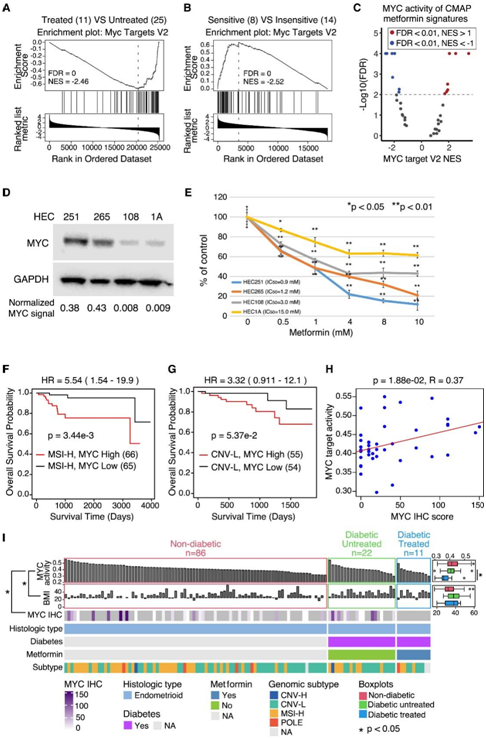
图4. 二甲双胍可能针对 EC 中的 MYC 。
(A) MYC Targets V2 富集图来自通路分析,比较独立队列中接受二甲双胍治疗与未治疗的 2 型糖尿病 (T2D) 患者。(B) 路径分析中的 MYC Targets V2 富集图,比较来自 DepMap 的二甲双胍敏感与不敏感 EC 细胞系。(C) CMAP 二甲双胍治疗特征的 MYC Targets V2 通路评分(x 轴)与 -log10(FDR)(y 轴)的火山图。(D) 免疫印迹显示四种 EC 细胞系中 MYC 的表达。(E) 用浓度递增的二甲双胍处理的 EC 细胞系的剂量反应曲线(x 轴)。(F-G) 具有高和低 MYC 活性的 TCGA MSI-H和 CNV-L肿瘤的生存分析。(H) MYC 活性(y 轴)与 MYC IHC 评分(x 轴)的散点图。(I) 独立队列中所有子宫内膜样肿瘤的热图,按 MYC 活性排序(上),并按糖尿病和二甲双胍治疗状态分组。箱线图(右)比较糖尿病/治疗组的 MYC 活性(上)和 BMI(下)。
05
CTNNB1热点突变(hotpot mutations)抑制 DKK 诱导的降解
CTNNB1基因第3外显子的热点突变(β-catenin)在CNV-L肿瘤中显著富集,是肿瘤发生的潜在驱动基因(图5A)。CTNNB1热点突变导致Wnt-β-catenin信号在RNA和蛋白质水平上显著上调,并下调几种免疫相关通路(图5B)。虽然Wnt通路抑制因子DKK4是上调最显著的蛋白,但在CTNNB1热点突变的肿瘤中,包括 CTNNB1 和 LEF1 在内的 Wnt 信号蛋白上调(图5C)。由于CTNNB1热点突变发生在关键磷酸化位点或邻近氨基酸,因此它们可能会阻止磷酸化依赖性 β-catenin 降解。S45位点磷酸化在热点突变的肿瘤中显著下调,而远离热点突变的磷酸位点大多上调(图5D)。他们还发现,在具有 CTNNB1热点突变的肿瘤中,细胞增殖和转运蛋白活性显著上调,整体免疫评分下调。
为了筛选潜在的蛋白质生物标志物,他们使用探索性队列的蛋白质组数据来训练回归模型,并使用独立队列测试其性能。使用CNV-L肿瘤和Wnt蛋白的模型表现最佳(AUC = 0.99)(图 5F-G)。他们验证了热点突变肿瘤中蛋白水平和MYC活性的升高。
综上所述,这些的结果表明,CTNNB1热点突变可阻断磷酸化诱导的β-catenin降解,可能使Wnt-FZD拮抗剂对有热点突变的内皮细胞无效(图5E),并且基于蛋白质的检测(如IHC)可用于检测热点突变状态。
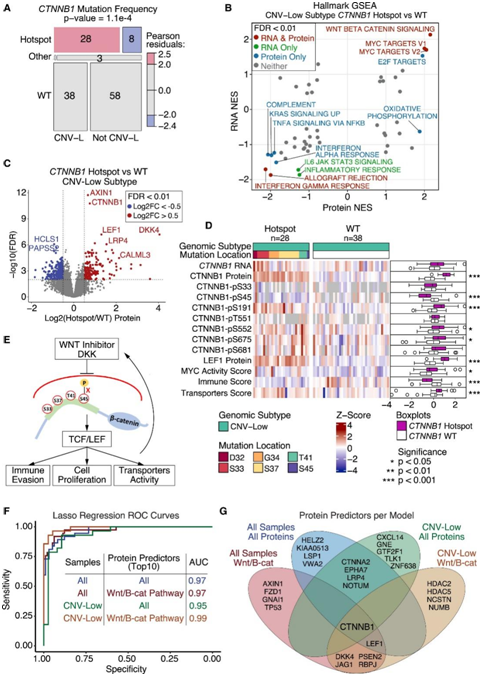
图5. CTNNB1热点突变阻断DKK诱导的降解。
(A) Mosaic图显示CNV-L 肿瘤与独立队列中所有其他肿瘤的CTNNB1突变分布。(B) 比较独立队列中CTNNB1热点突变体与 WT CNV-L 肿瘤在蛋白质(x 轴)和 RNA(y 轴)水平上的通路标准化富集分数 (NES) 散点图。(C) CTNNB1热点和 WT CNV-L 肿瘤之间的蛋白质 log2 差异倍数(x 轴)与通过 Student t 检验确定的 -log10 FDR(y 轴)的火山图。(D) 热图显示了具有和不具有CTNNB1热点突变的 CNV-L 肿瘤中 CTNNB1、LEF1 蛋白、MYC 活性评分、免疫评分和转运蛋白评分的 mRNA、蛋白质和磷蛋白值。(E) 示意图描绘了CTNNB1中热点突变的拟议下游影响。(F) 使用探索性队列蛋白质数据作为训练和独立蛋白质数据作为测试来预测CTNNB1热点突变状态的 Lasso 回归模型的 ROC 曲线。(G) 维恩图显示每个模型通过回归分析选择的前 10 个蛋白质。
06
使用组织病理学切片的深度学习模型预测亚型和突变状态
接下来,他们建立了卷积神经网络模型,以预测 EC 的分子亚型、组织学亚型和常见突变,并在独立队列中对其进行了验证(图 6 A),能够成功区分 CNV-H 和非 CNV-H 样本(图 6B-C)。
CNV 增加是癌症的一个标志,与较差的结果、增加的免疫逃避和对免疫疗法的反应降低相关。虽然CNV- H亚型显示出最高浓度的CNV,但在所有EC亚型中都有染色体特异性改变的模式,例如染色体1q的增益,这是EC中最常见的臂水平的CNV(图6D)。具有染色体1q增益的肿瘤,其总体免疫评分、CD8+幼稚T细胞评分、骨髓树突状细胞评分和微环境评分显著降低(图6E)。使用糖蛋白质组学数据,他们观察到肿瘤中聚合免疫球蛋白受体 (PIGR) 和其他糖基化蛋白的糖肽上调伴随着染色体1q增益(图 6 F)。他们观察到PARP1拷贝数、RNA、蛋白质、磷酸化和乙酰化水平增加,不仅在1q增益的肿瘤中,而且在PARP1局灶性扩增的肿瘤中(图6G)。为了检测PARP1扩增和对PARP抑制剂(PARP-i)的药物反应是否有相关性,他们使用DepMap观察到有PARP1扩增的EC细胞系对PARP-i奥拉帕利的敏感性增加(图6H)。PARP1的表达可能是EC肿瘤的进化优势,并可能成为PARP-i治疗的潜在生物标志物。
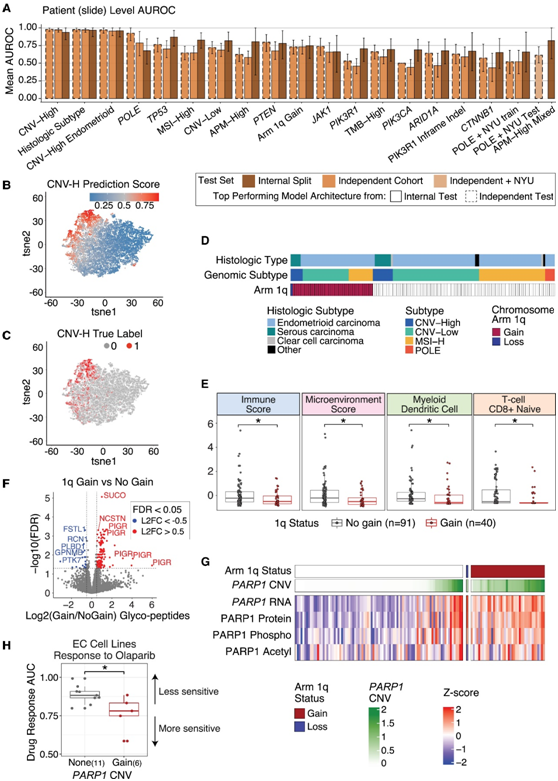
图6. 深度学习模型成功对分子特征进行分类。
(A) 条形图显示来自内部训练数据分割测试(在 TCGA 和探索性队列上训练)和独立测试(在独立队列加上 NYU 队列上进行 POLE 预测测试)的每个模型的平均 AUROC。(B-C) tSNE 图,其中每个点都是一个图块,由预测的 CNV-H 分数和真实的 CNV-H 特征着色。(D) 独立队列中所有肿瘤的染色体 1q 拷贝数状态分布,按基因组和组织学亚型分组。(E) xCell 免疫评分(y 轴)的箱线图,比较有 1q 增益与无增益(x 轴)的肿瘤。(F) 具有染色体1q 增益与无增益的肿瘤中差异表达糖肽的火山图。(G)有和没有 1q 增益的样本中PARP1多组学水平的热图。(H) 具有和不具有 PARP1扩增的 DepMap EC 细胞系中奥拉帕尼(PARP 抑制剂)响应的箱线图。
07
多组学聚类揭示了富含CTNNB1热点突变的 CNV-L 亚组和 1q 增益 MSI-H 亚组
多组学聚类揭示了富含CTNNB1热点突变的 CNV-L 亚组和染色体1q 增益 MSI-H 亚组(图7A)。所有CNV-H肿瘤都属于聚类1,其中TP53突变肿瘤显著富集并且具有增殖特征(图7B)。聚类2 和聚类3 富含 CNV-L 亚型,聚类3 的特征为CTNNB1热点突变、arm 1q 增益、上调转运蛋白活性并下调免疫通路。聚类 4 富含 MSI-H和 TMB-H,并且包括大多数 POLE 肿瘤。聚类2 和聚类3 具有互补的通路特征,聚类2 显示免疫相关通路的上调。除了 mTOR 和 TNFα 信号传导之外,聚类4 还增强了细胞周期、代谢和 DNA 损伤相关通路的活性。三个聚类之间仅 MSI-H 样本的比较揭示了聚类 3 中 1q 增益的富集和聚类 4 中ELMSAN1突变的富集,表明 MSI-H 亚型内存在亚组。
糖肽水平的无监督 NMF 聚类将肿瘤分为四种亚型(图 7 C)。聚类1 和聚类3 主要与复杂聚糖唾液酸 (Sia)和低聚甘露糖 (HM) 相关,聚类 2 与 Sia 相关,聚类4 与低聚甘露糖 (HM) 相关。染色体1q增益在聚类1中最常见,TP53突变在聚类2和聚类4中频繁,CTNNB1突变在聚类2中罕见。与寡糖前体合成相关的蛋白质在所有四个聚类中均下调(图 7D)。
聚焦于肿瘤和正常样本之间的 PI3K-AKT 通路差异(图 7E),他们观察到来自 7 种激酶的 15 种糖肽。其中大多数在肿瘤中表达上调,包括 FLT1 的多个糖基化位点。在 EGFR 上观察到显著下调的寡甘露糖结构,这表明肿瘤样品中 EGFR 的糖基化可能发生改变,这可能会影响 EGFR 在肿瘤中的功能。
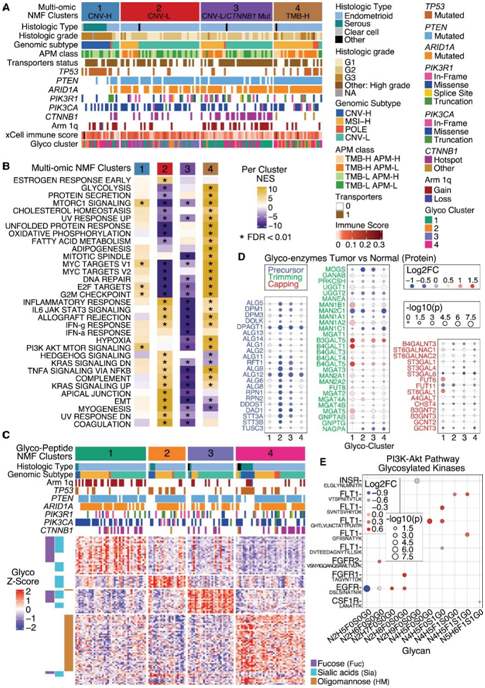
图7. 多组学和糖蛋白组 NMF 聚类将样品分成 4 个聚类。
(A) 独立队列中所有肿瘤的热图,由多组学 NMF 聚类分隔。(B) 每个聚类的平均 ssGSEA 通路富集热图。(C) 所有独立队列肿瘤的糖肽水平热图。(D) 肿瘤与正常糖聚类之间的糖酶水平点图。(E) 通过聚糖(x 轴)和相应的肽(y 轴)比较肿瘤和正常样品的 PI3K-AKT 通路中的糖基化激酶的点图。
+ + + + + + + + + + +
结 论
本项研究使用 10 个不同的组学平台对包含 138 个肿瘤和 20 个富集正常组织的子宫内膜癌 (EC) 队列进行了表征。两种肽的靶向定量可以预测抗原加工和呈递机制的活性,并可以为患者选择免疫治疗提供信息。患者和细胞系中 MYC 活性与二甲双胍治疗之间的关联分析表明,二甲双胍治疗对于 MYC 活性升高的非糖尿病患者具有潜在作用。PIK3R1插入缺失与 AKT 磷酸化升高和对 AKT 抑制剂敏感性增加相关。CTNNB1热点突变集中在介导 pS45 诱导的 β-catenin 降解的磷酸化位点附近,这可能导致 Wnt-FZD 拮抗剂无效。深度学习模型可以根据组织病理学图像准确预测 EC 亚型和突变,这可能有助于快速诊断。总体而言,本研究确定了可以进一步研究的分子和影像标志物,以指导患者分层,从而更精确地治疗 EC。
+ + + + +



