

 English
English文献解读|Cancer Cell(44.5):不同种族和暴露水平的肺腺癌综合分析
✦ +
+
论文ID
原名:Integrative analysis of lung adenocarcinoma across diverse ethnicities and exposures
译名:不同种族和暴露水平的肺腺癌综合分析
期刊:Cancer Cell
影响因子:44.5
发表时间:2025.07.30
DOI号:10.1016/j.ccell.2025.07.011
背 景
肺腺癌 (LUAD) 是非小细胞肺癌 (NSCLC) 中最常见的亚型,其发病率和疾病进展存在显著的地理和流行病学差异,是全球面临的重大健康挑战。虽然靶向治疗和诊断技术取得了进展,LUAD 的死亡率仍然很高,这表明有必要深入了解不同人群中 LUAD 的分子基础。将蛋白质组学、翻译后修饰与基因组学相结合的蛋白质组学研究可以识别临床分层和致癌机制,但其效力不足以探究种族、吸烟、环境暴露或性别对这种异质性疾病的影响。
实验设计
结 果
01

ICPC-CPTAC LUAD 队列的蛋白质组学特征
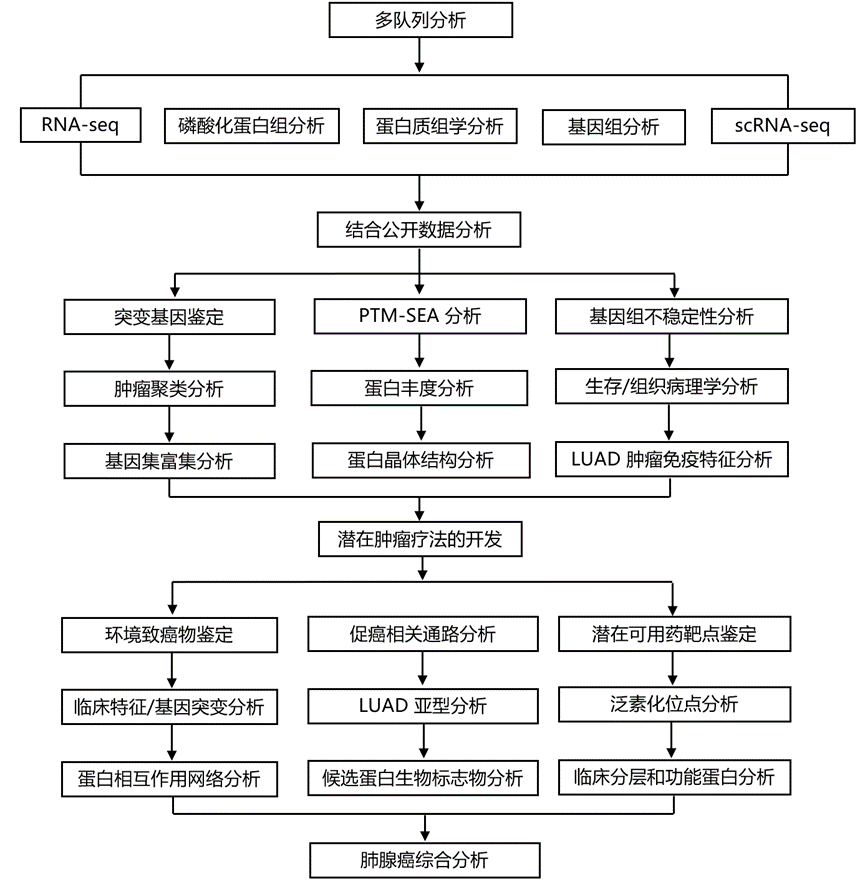
美国国家癌症研究所 (NCI) 临床蛋白质组肿瘤分析联盟 (CPTAC) 和国际癌症蛋白质组联盟 (TW-ICPC) 下的台湾癌症登月团队此前曾对具有不同人口统计数据的不同 LUAD 患者队列进行了单独分析。已发布的CPTAC 队列(“CPTAC.A”)分别包括富含KRAS或EGFR突变的西方和亚洲吸烟者、曾经吸烟者和从不吸烟者。相比之下,TW-ICPC 研究侧重于台湾地区从未吸烟的 LUAD 病例,主要为肿瘤携带EGFR突变的女性(“ICPC.A”)。研究团队结合这些已发表的队列以及来自每个团队的其他病例(“CPTAC.B”和“ICPC.B”)进行了联合分析。这个规模庞大且多样化的 LUAD 蛋白质组学队列包含 406 例 LUAD 肿瘤和 388 例匹配的正常邻近组织 (NAT)(图 1 A)。
所有队列产生的多组学数据包括基因组数据、转录组学分析(RNA-seq)数据、蛋白质组学和磷酸化蛋白质组学分析数据(图 1 A)。Oncoplot 强调了涉及RAS/RTK通路和广泛共突变的已知致癌改变的预期相互排斥性,包括肿瘤抑制基因,如TP53、STK11、RBM10和CDKN2A(图 1B)。基于细胞表型的细胞起源分析鉴定出 AT2 和 Club 细胞在EGFR、RAS和RAF家族突变肿瘤中富集,与一小部分肺鳞状细胞癌 (LSCC) 样本形成对比,这些样本显示出基底细胞的富集,正如预期的那样(图 1C)。有趣的是,与谱系特异性或细胞类型富集的对应物相比,无法确定细胞起源的 RTK 改变细胞系对靶向疗法的反应较差。治疗反应的这种异质性强调了细胞转录状态的潜在差异。
基于非负矩阵分解(NMF)的多组学聚类定义了四个肿瘤聚类(图1D),其临床生物学注释和分子特征富集差异(图1E-F)。第一类"不稳定增殖型(UP)"样本表现出基因组不稳定性证据及与高增殖一致的特征;第二类"静息EGFR型(QE)"显示多个关键致癌通路的下调及EGFR突变富集;第三类"免疫活跃KRAS型(IAK)"呈现免疫信号通路上调及KRAS突变富集;第四类"稳定早期型(SES)"则显著富集早期阶段且基因组稳定的肿瘤。本研究队列的多组学聚类结果与TCGA LUAD研究的转录组亚型(分别为LUAD亚型和TCGA亚型)及CPTAC既往多组学聚类(CPTAC.A C1-C4)进行了对比(图1D)。虽然ICPC-CPTAC队列在人口统计学上具有独特性,但部分生物学特征在不同队列间保持保守:例如IAK聚类与LUAD亚型S3、TCGA亚型proximal_inflammatory高度重合,且与CPTAC.A C1强相关,均显示免疫信号通路的显著正富集;而SES聚类则与LUAD亚型S5、TCGA亚型terminal_respiratory_unit富集一致,并与CPTAC.A C4强相关,均表现为细胞周期相关通路的负富集。
基于NMF的聚类也在个体组学空间中进行,包括RNA、蛋白质、磷酸化蛋白和基因水平的CNA。蛋白质组聚类值得关注,它将患者分为三个聚类(C1-C3),每个聚类与独特的临床和表型特征相关,包括在体能状态和预后较差的患者中,基因组不稳定、免疫“热”肿瘤在蛋白质组聚类2中的富集(图1 D)。

图1. CPTAC 和 ICPC 队列的蛋白质组学图谱。
(A) 左图:纳入样本及组学数据汇总。样本数量按肿瘤和正常癌旁组织 (NAT) 分层。右图:本研究中 406 名患者中选定注释的分布情况。(B)406例患者队列中目标突变的Oncoplot图。(C) 对 402 例 RNA-seq通过质量控制的肿瘤样本进行细胞来源分析。(D) 对 383 个肿瘤样本进行多组学 NMF 聚类,并整合多组学数据。 (E) 临床注释在每个多组学聚类中显著富集。(F) 四个多组学聚类的基因集富集分析(GSEA)。
02
RBM10突变和ALK融合等不太常见的遗传事件的生物学影响
合并后的 LUAD 队列规模庞大,使研究团队能够研究相对少见的共突变和驱动事件,包括RBM10突变和ALK融合的蛋白质组学后果。与先前在规模较小且多样性较低的队列中进行的研究不同,RBM10突变与TP53突变并不互斥,也与男性性别无关,但截短突变与蛋白质组学驱动的剪接体相关通路下调相关(图 2 A)。他们观察到剪接激酶 CLK1-3 和 SPRK 的调节以及磷酸化驱动的 EGFR、NOTCH 和 KIT 受体信号转导上调(图 2 B)。RBM10突变样本中剪接体的下调与许多癌症中的上调形成对比,并且可能在EGFR突变的背景下赋予对剪接体抑制的依赖性相关敏感性。与观察到的代谢改变一致(图 2 A),催化糖酵解第一步的酶己糖激酶 2(HK2)在RBM10/EGFR共突变样本中显著上调(图 2 C),而 SF3B1 的蛋白质表达基本没有变化,这表明对剪接体抑制的敏感性可能不是由于 SF3B1 复合物的活性。最后,他们观察到RBM10共突变的EGFR-L858R亚群中中性粒细胞脱颗粒 (ND) 特征显著上调。在 RNA 和蛋白质水平上均观察到的 ND 在另一个患者数据集和RBM10敲除小鼠模型中得到了验证,为共突变的EGFR-L858R和RBM10 LUAD 中的从头ND 上调提供了进一步的证据。RBM10 缺陷与对 EGFR 抑制剂疗法的耐药性有关,本研究的数据表明RBM10改变的功能性后果扩展到免疫微环境,对细胞毒性和免疫检查点抑制剂疗法具有潜在影响。
与RBM10、EGFR和KRAS突变不同,与受体酪氨酸激酶ALK相关的融合相对少见,但可作为LUAD驱动事件的靶点,主要见于非吸烟者。由于在ALK融合驱动的LUAD中,组成性激活的ALK在组织水平上异位,在细胞水平上错误定位,磷酸化蛋白质组学为揭示致癌生物学机制提供了独特的机会。他们此前在ALK融合阳性的CPTAC.A LUAD样本中报告了SND1、EML4、HDLBP、LHDA、ARHGEF5和PIK3R1等蛋白质上特定酪氨酸(Y)残基的磷酸化增加,在合并的LUAD队列中也得到了证实(图2 D)。为了评估这些观察结果的功能意义,他们在具有ALK融合或激酶驱动突变(FGFR/2、KRAS和PIK3CA)的细胞系中进行了激酶抑制实验,并通过质谱法检查了 ALK 相关的 pY 位点。用 ALK 抑制剂处理ALK融合驱动的细胞系,而不是用其他激酶驱动/抑制剂匹配的治疗方法,导致 ALK 相关 pY 显著下调(图2E),支持它们对 ALK 的依赖性。使用磷酸化形式的肽作为诱饵,在来自EML4-ALK融合驱动的细胞系的裂解物中进行肽 pull-down 分析,导致 ALK 以及许多是 ALK 相互作用因子的蛋白质(例如 PTPN11 和 SND1)的恢复(图 2 F)。总之,这些实验证实ALK融合促进了一系列明确的磷酸化事件,从而促进了关键蛋白质的相互作用。
蛋白质翻译后修饰(PTM)与突变类似,可显著增加蛋白质形式的多样性并影响癌症生物学。聚集性PTM可能产生更强的生物学效应,或指示功能关键位点,尤其在蛋白质三维结构中的共定位位点。为识别蛋白质三维结构内相关性PTM聚类,他们采用CLUMPS-PTM方法进行分析。在肿瘤vs癌旁组织、吸烟者vs非吸烟者、EGFR突变vs野生型肿瘤的配对比较中(图2G),32种蛋白质的磷酸化、乙酰化或泛素化位点呈现显著聚集。在EGFR突变与野生型肿瘤对比中,小热休克蛋白HSPB1成为最显著差异蛋白(图2H)。HSPB1 S78和S82位点的差异化磷酸化可促进其与AKT1等蛋白的物理相互作用,导致后者磷酸化并通过促进中性粒细胞凋亡触发抗炎反应。与此一致,在嗜酸性粒细胞/内皮细胞(E/E)免疫亚型(该亚型中性粒细胞相对缺失且EGFR突变富集)中观察到HSPB1 S82/S83位点磷酸化水平升高。同时,EGFR突变样本中AKT1蛋白表达更高(图2H)。在明确抑癌基因及组蛋白乙酰转移酶EP300中观察到乙酰化位点聚类。肿瘤vs NAT比较中,已知EP300激活位点K1546、K1549、K1554和K1555的乙酰化水平升高,这与CBP/EP300调控的底物H2A和H2B的N端乙酰化水平升高相关,后者是活性增强子的标志物——增强子异常激活驱动癌症进展的机制已在多种癌症中得到证实。蛋白质乙酰化对癌症代谢同样重要,如MDH1的差异化乙酰化通过产生NAD+和苹果酸等代谢物支持肿瘤糖酵解。已知MDH1的K328、K329和K335是泛素化位点,乙酰化可稳定并增强该酶活性。他们在调控DNA双链断裂(DSB)修复并通过ATM-ESCO2-SMC3轴维持基因组完整性的SMC3上发现乙酰化位点聚类,其主效乙酰化位点K105/K106与基因组不稳定性显著相关。SMC1A复合体成员也呈现类似乙酰化位点特异性关联,进一步凸显PTM分析独有的功能特性。最后,在20S核心蛋白酶体亚基PSMA5的蛋白酶体结构域内直接观察到泛素化位点聚类(图2J)。K187、K192、K196和K203位点的泛素化会抑制蛋白酶体活性。PSMA5可促进肿瘤进展并与不良预后相关,其在基于NMF蛋白质聚类C2组(晚期肿瘤患者比例最高且生存较差)中表达显著升高。
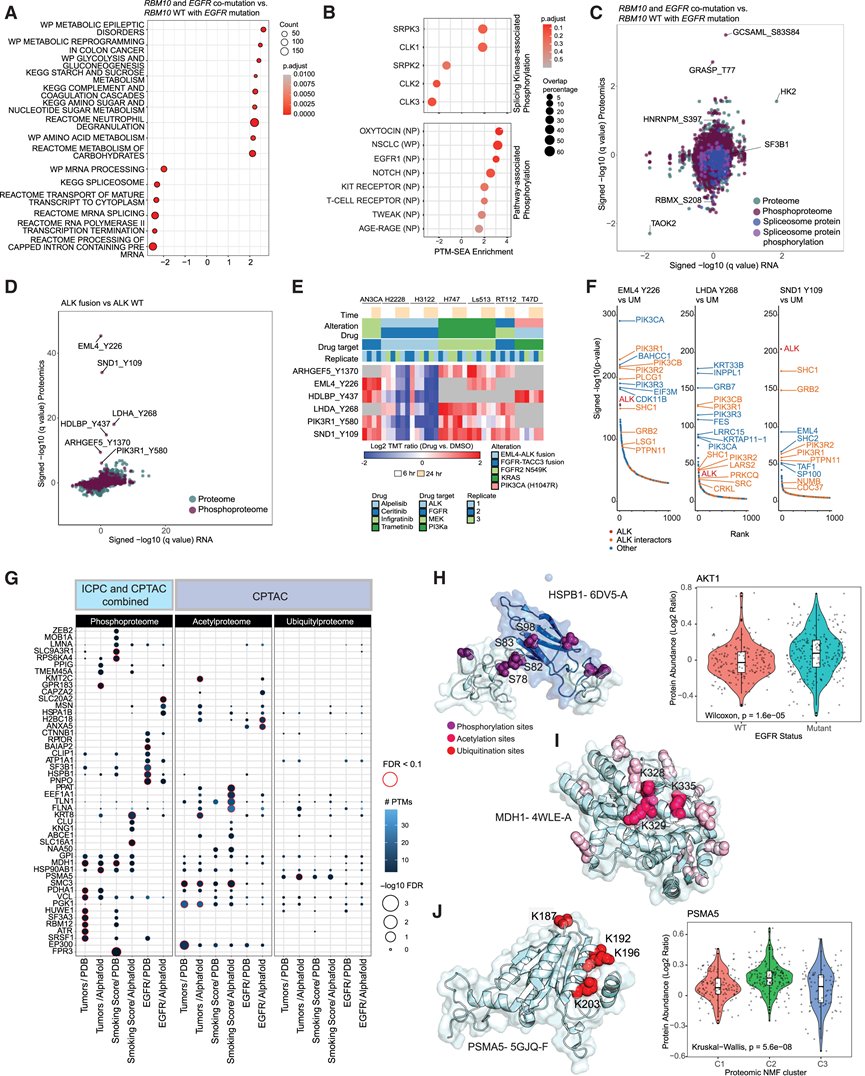
图2. 突变和 PTM 对下游信号和结构的影响。
(A) GSEA 蛋白质水平差异表达分析结果。(B)翻译后修饰-特征富集分析 (PTM-SEA) 显示剪接激酶和通路相关的磷酸化富集。(C) 在蛋白质组和磷酸化蛋白质组水平上,EGFR突变背景下RBM10突变体和 WT 肿瘤之间的差异表达特征。(D) ALK融合肿瘤和 WT 肿瘤在蛋白质组和磷酸化蛋白质组水平上的差异特征。(E) 候选 pY 位点及其在用驱动匹配激酶抑制剂处理的一系列细胞系中的丰度。(F) 排序图显示候选 pY 肽与未修饰肽 Pull-down 肽段的蛋白质丰度。(G) 气泡图表示 CLUMPS-PTM 结果。(H)HSPB1磷酸化聚类的3D晶体结构。(I) MDH1乙酰化聚类的3D晶体结构。(J) PSMA5 泛素化聚类的 3D 晶体结构。
03
基因组连续性与断裂强度聚类相结合对 LUAD 具有预测作用
染色体不稳定性(CIN)是癌症发生与进展的重要特征。与既往研究一致,他们发现常用CIN指标(如加权基因组不稳定性指数wGII)无法有效区分LUAD预后。这些指标主要量化基因组失衡(非整倍性)而非片段化程度(连续性),后者反映在基因组片段长度上。他们通过片段长度分布评估基因组连续性,并以该分布SegLen-Q3作为长片段存在的替代指标,据此将样本分为完全稳定、部分稳定或不稳定三类(图3A)。基于SegLen-Q3的分类与总生存期(OS)显著相关(图3B),该关联在多种癌症的独立队列中得到验证。不稳定肿瘤呈现TERT、NKX2-1和MYC的局灶性扩增及预期结构变异增加(图3C),其中TERT扩增同样与OS相关。
基因组片段长度的分布不仅取决于样本中基因组断裂点的数量,还取决于这些断裂点的全基因组分布模式——即断裂点倾向于聚集(形成少量长片段和大量短片段)还是分散(形成多数中等长度片段)。基于此,他们应用空间点过程理论开发了断裂强度聚类(BIC)指标,分别评估基因组断裂的强度(int)和聚集性(clust)。根据这两个参数对肿瘤进行分类(图3D),可识别出生存期极佳(BIC-contiguous)、典型(BIC-fragmented)和较差(BIC-intense)的患者分层(图3E)。BIC分类能独特识别出显著断裂集中于小基因组区域的肿瘤。
NKX2-1的局灶性扩增主要局限于BIC-intense型肿瘤(图3C),且该组肿瘤的扩增比例显著高于其他BIC分组(图3F)。扩增伴随NKX2-1 RNA和蛋白表达水平显著升高。在部分BIC-intense和BIC-fragmented型肿瘤中,他们还发现NKX2-1的完全转录沉默(图3F),这与片段化型NKX2-1沉默肿瘤中启动子甲基化水平升高相关。值得注意的是,NKX2-1扩增和沉默均与更高增殖活性相关(图3G)。缺失NKX2-1的肿瘤同时丧失了肺泡细胞特征,这通过转录程序推断和标志基因Napsin A(NAPSA)表达得到证实(图3H)。相反,NKX2-1扩增肿瘤在维持谱系特征和NAPSA表达的同时,具有高增殖指数(图3G)。NKX2-1沉默肿瘤中,癌症睾丸抗原(如TESMIN、MAGEA和MAGEA12)以及与LUAD进展和鳞状分化相关的标志物(包括KRT6A和KRT16)显著上调。
靶向染色体不稳定肿瘤依赖性特征的最新疗法,为开发稳健的不稳定性生物标志物提供了理论依据。通过比较片段化与其他癌症基因组(图3I),他们鉴定出真性癌基因IGF2BP3作为基因组不稳定相关的主要候选生物标志物。他们优化了免疫组化(IHC)检测方法,结果显示IGF2BP3在不同基因组不稳定性水平样本中呈现差异化染色模式(图3J),完全稳定样本中未见表达。该IHC方法在包含初治患者独立样本(78例LUAD、92例LSCC、69例转移性LUAD及60例配对NAT)的组织芯片上得到验证。IGF2BP3在正常肺组织中无表达,在部分原发LUAD和LSCC中升高,在几乎所有转移性LUAD(图3K)以及ICGC/PCAWG队列的不稳定LUAD中显著高表达(图3L)。实验诱导基因组不稳定可导致不稳定克隆中IGF2BP3蛋白水平升高,提示其作为染色体不稳定动态标志物的潜力。IGF2BP3的均匀胞质染色与细胞周期依赖性Ki67蛋白的斑片状染色形成鲜明对比。
染色体不稳定肿瘤通过多种机制逃避免疫监视,他们因此探究IGF2BP3能否作为免疫检查点抑制剂(ICI)治疗的预测标志物。一项比较ICI与多西他赛治疗NSCLC的随机试验数据显示,无论PD-L1(CD274)表达水平如何(图3N),高IGF2BP3蛋白表达均与ICI治疗较差预后相关(图3M),而与多西他赛疗效无关。IGF2BP3蛋白的高表达可能由启动子去甲基化触发(图3O)。最具侵袭性的BIC-intense肿瘤以TP53/EGFR突变、全基因组倍增(WGD)和TERT扩增为特征,与临床分期或吸烟状态仅呈弱相关,且KRAS突变较少。
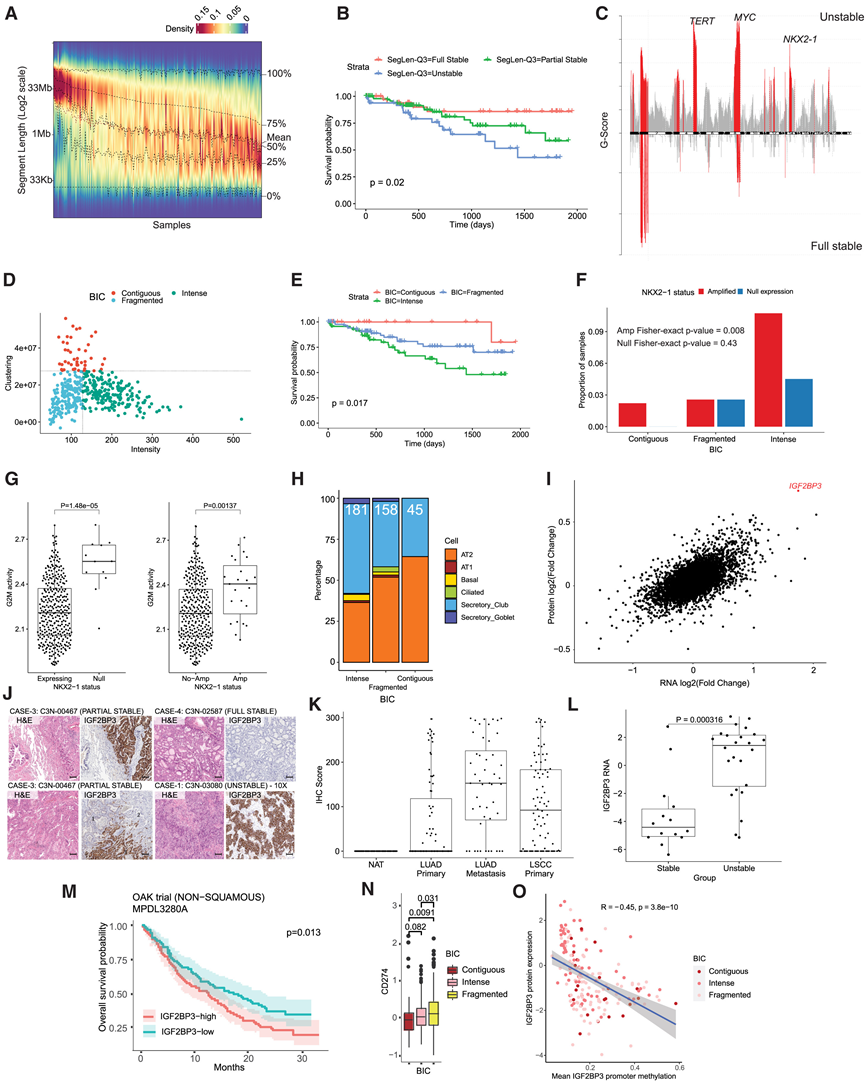
图3. LUAD 中的基因组不稳定性及其影响。
(A) 每个样本的 CNV 片段长度分布。(B) Kaplan-Meier (KM) 生存曲线显示患者的总体生存率。(C) 不稳定(顶部)和完全稳定(底部)肿瘤中反复扩增区域的比较。(D)根据断点强度(x 轴)和聚类得分(y 轴)对肿瘤进行分类。(E) KM生存曲线显示患者的总生存率,按BIC分层。(F) NKX2-1扩增(红色)和 NKX2-1 表达缺失(蓝色)的肿瘤比例。(G) 按NKX2-1扩增(左)和表达(右)状态分层的肿瘤增殖活性。(H) 按 BIC 组分层的肿瘤转录状态。(I) 通过 RNA(x 轴)和蛋白质(y 轴)水平比较 BIC 强度与其他肿瘤的差异表达分析。(J) IGF2BP3 IHC 和 H&E 染色显示。(K) 对3例TMA患者的独立验证显示,转移性LUAD中的IGF2BP3表达高于原发性LUAD和原发性LSCC。(L) ICGC/PCAWG 中 LUAD 患者的IGF2BP3 (RNA)表达,按不稳定性分层。(M)KM 生存曲线显示了用抗 PDL1 抗体 MPDL3280A 治疗的非鳞状患者的总体生存率。(N) 按 BIC 状态分层的 CPTAC 队列中的 PD-L1 (CD274) 蛋白表达。(O) IGF2BP3 蛋白表达与平均 IGF2BP3 启动子甲基化的相关性。
04
热和冷 LUAD 肿瘤中的差异免疫信号以及推断的转录因子和激酶活性
免疫调节治疗的成功已经改变了肿瘤治疗,但许多癌症,包括EGFR突变的 LUAD,仍然难以通过免疫疗法治愈。为了加深对免疫背景和免疫生物学的理解,他们通过整合 406 个肿瘤和 388 个匹配的 NAT 的蛋白质组学和基因表达数据进行了反卷积分析。使用基于泛癌症 RNA 和蛋白质组学数据的分类器,他们将 LUAD 样本分为 5 种免疫亚型:CD8+/IFNG+、嗜酸性粒细胞/内皮细胞 (E/E)、成纤维细胞/TGFBeta、CD8-/IFNG+ 和 CD8-/IFNG-(图 4 A)。正如预期的那样,CD8+/IFNG+ 中的样本富含 CD8 +和 CD4 + T 细胞,并显示干扰素γ信号传导的激活,这也在 CD8-/IFNG+ 中观察到(图 4B)。EGFR 信号通路在 CD8+/IFNG+ 中上调(图4B)。E/E 中的样本富含成纤维细胞,反映了它们的基质细胞含量,并且与低吸烟暴露评分和低肿瘤突变负荷 (TMB) 相关。该亚型还以“MAPK1/ERK2 活化”特征和下游“RSK 活化”通路的蛋白质组学上调为特征,这是肺癌的一个有希望的治疗靶点(图 4B)。CD8-/IFNG- 和 CD8-/IFNG+ 均与“MYC 靶点”、“E2F 靶点”、“DNA 修复”和“细胞周期检查点”的上调有关,表明这两种免疫亚型中细胞周期相关通路的激活。
根据 CNA 和基因表达数据估算的肿瘤细胞百分比与免疫细胞分数呈负相关,在高免疫原性 CD8+/IFNG+ 肿瘤中降低(图 4 A)。免疫亚型与基于 RNA 的 LUAD 分类相符, CD8+/IFNG+ 肿瘤在“免疫活性 KRAS (IAK)”聚类 (S3) 中富集,在 RNA S5 肿瘤中富集 E/E(图 4 A)。他们观察到 CD8+/IFNG+ 在 C2 蛋白质组聚类中富集(图 4 A),提示某些 C2 肿瘤可能受益于免疫疗法。
NAT 的反卷积分析表明,与肿瘤组织相比,髓系细胞(如巨噬细胞和 NK 细胞)的浸润更高。细胞类型分数和突变谱之间的关联分析显示,TP53突变与 CD8 + T 细胞、中性粒细胞和树突状细胞 (DC) 的存在较高相关,具有潜在的治疗相关性,因为之前有报道称 TP53 突变肿瘤中的 CD8 + T细胞在免疫治疗反应中具有益处。接下来,利用磷酸化蛋白质组学数据,他们进行了激酶富集分析(KEA3)以得出每位患者的激酶活化评分,并与免疫亚型进行了关联分析。他们观察到 CD8+/IFNG+ 肿瘤中酪氨酸激酶(如 HCK、CSK 和 BTK) 的活化程度更高(图 4 C)。酪氨酸激酶是免疫系统中的信号调节器,其中 Src 家族激酶 (如 HCK)在白细胞中更活跃。另一方面,CD8-/IFNG+ 和 CD8-/IFNG- 与细胞周期相关激酶(如 CHEK2、CSNK2A2 和 CDK9)的活化有关,反映了较冷的肿瘤微环境。与通路分析一致,他们还观察到 E/E 亚型中 MAPK1 (ERK2) 和 RPS6KA1 (RSK) 的活化。
由于蛋白激酶作为细胞信号传导开关,调节细胞内程序,例如转录和翻译,他们旨在鉴定在热肿瘤(CD8+/IFNG+)和冷肿瘤(CD8-/IFNG-)中差异激活的激酶下游更完整的细胞信号传导通路。为此,他们评估了推断的激酶和转录因子 (TF) 的共活性,分别使用 TF 和激酶富集分析对每位患者的差异表达 mRNA 和磷酸化蛋白进行了分析。这使他们能够探究在热肿瘤和冷肿瘤中正向或负向调控 TF 模块的激酶模块。七个激酶模块中有两个包含免疫相关的酪氨酸激酶,包括 SYK、LYN、LCK 和 FYN,五个 TF 模块中有两个富含免疫相关的 TF,例如 RUNX3、FOXP3 和 STAT4,这些 TF 在冷性肿瘤中似乎下调。相反,包含 CDK1/2 和 AURKB 的细胞周期相关激酶模块仅与热肿瘤中免疫 TF 模块的上调相关。另一个富含与粘着斑相关的激酶的模块,包括 MAPK8/12、MAP2K1、PTK6、TESK2 和 IGF1R,与前面提到的免疫相关 TF 模块相反,在热肿瘤中下调(图 4D)。在KRAS/STK11共突变的 LUAD 小鼠模型中,抑制粘着斑与免疫浸润增加和冷热转变相关,这表明,靶向该模块中的激酶可能会使肿瘤微环境转向更允许的表型。使用 LINCS L1000 数据集验证了这种关联,该数据集分析了在 CRISPR 敲除约 8000 个单基因和用小分子和药物进行约 30000 次化学扰动后人类癌细胞系中基因表达的变化。具体来说,他们检测了粘着斑模块中激酶 CRISPR 敲除后上调的基因,并证实这组上调的基因与 Reactome 先天免疫途径(先天免疫系统 R-HSA-168249)和从已发表的 ChIP-seq 研究和其他来源获得的 STAT4 下游基因显著重叠(图 4D)。
根据RNA 和蛋白质组数据,他们发现“MTOR 信号传导”、“癌症中的 WNT 信号传导”和多种代谢途径在冷性肿瘤的肿瘤细胞中上调(图 4 E),而根据蛋白质组数据,“先天免疫系统”和“Notch 信号传导”在热肿瘤的肿瘤细胞中上调。WNT 信号传导不仅与肿瘤发展有关,还与肿瘤细胞逃避宿主免疫反应的能力有关。为了验证热和冷肿瘤肿瘤细胞中这种通路的激活,他们利用了一个单细胞转录组分析(scRNA-seq)数据库,该数据库包含 33 名免疫含量低(冷)患者和 40 名免疫含量高(热)患者。scRNA-seq 数据发现,冷性肿瘤细胞中多种代谢通路和“MTOR 信号”上调(图4E)。另一方面,scRNA-seq 数据证实,热性肿瘤细胞中仅“先天免疫反应”上调。
分析了蛋白质糖基化(通常富含膜和细胞外基质蛋白)与 LUAD 免疫亚型的关联,发现几种蛋白质的糖基化,包括 FKBP10和 PLOD2,它们之前已与 LUAD 进展相关并且是胶原 1 蛋白稳态网络的成员,在E/E 亚型中下调(图 4 F)。这可能与 E/E 亚型的中性粒细胞浸润较低和生存率更高有关(图 4 G-H),如前所述。这些发现与先前对 LUAD 生存率的研究一致并支持糖基化对亚型特异性结果的影响,值得进一步研究。
LUAD中STK11突变与疾病侵袭性强、预后较差相关,但其与免疫治疗疗效的关系存在争议。对STK11突变样本中显著异常特征的通路分析显示代谢通路失调。既往研究提示STK11突变与LUAD及泛癌免疫分析中的"冷肿瘤"微环境相关。然而在本跨国LUAD队列中,STK11突变仅与树突状细胞耗竭相关(图4I),而其他炎症性肿瘤微环境指标(如肿瘤细胞比例、免疫评分)无显著差异。STK11突变肿瘤在免疫亚型中的分布特征提示需深入研究其异质性,包括同时存在STK11与KEAP1突变的情况(既往研究显示这类LUAD具有独特性质)。蛋白质组聚类C1/C3组的STK11突变肿瘤与C2组存在显著差异:C1/C3组STK11突变肿瘤具有更低的wgII和TMB,且主要富集于S5 RNA亚型而非先前认为的主要S4亚型。先前报道的中性粒细胞脱颗粒特征(STK11突变LUAD的标志)仅在C2组STK11突变肿瘤中出现,C1/C3组未见此现象。
C1/C3与C2组STK11突变肿瘤在癌症相关分子异常分布上存在显著差异。C1/C3组并未显示更高的STK11/KEAP1共突变率,表明其差异机制与共突变无关。这类肿瘤呈现更高CD47表达及更稳定的基因组特征(图4J),表现为MKI67、CDK1、TOP2A和IGF2BP3蛋白表达下调(图4K)。在蛋白质组通路层面,这些肿瘤中性粒细胞脱颗粒活性降低,而异生物代谢、血红素代谢和活性氧通路则上调。NRF2通路活性在多个分子层面(包括PTM)增强(图4K)。低中性粒细胞脱颗粒的STK11突变肿瘤(C1/C3)具有铁死亡抵抗表型,该特征通过NEDD4样E3泛素连接酶(NEDD4L)上调和乳铁蛋白(LTF)下调得以强化——这种抑制铁死亡的新机制在转录组层面无法观测(图4K)。铁死亡保护基因NQO1是潜在治疗靶点。部分STK11突变肿瘤的聚类特异性差异也存在于同聚类野生型肿瘤中(如C2组普遍存在中性粒细胞脱颗粒和高IGF2BP3表达),而CD47、NEDD4L和UGDH的表达差异则与NRF2通路活性无关,为STK11突变特有。C1/C3组STK11突变肿瘤还表现出胃分化标志物(MUC5AC、GKN2、PGC和CTSE)表达及IGF2BP3/EZH2下调,这种异质性提示,针对STK11突变肿瘤可能需要进一步分层以挖掘治疗脆弱性
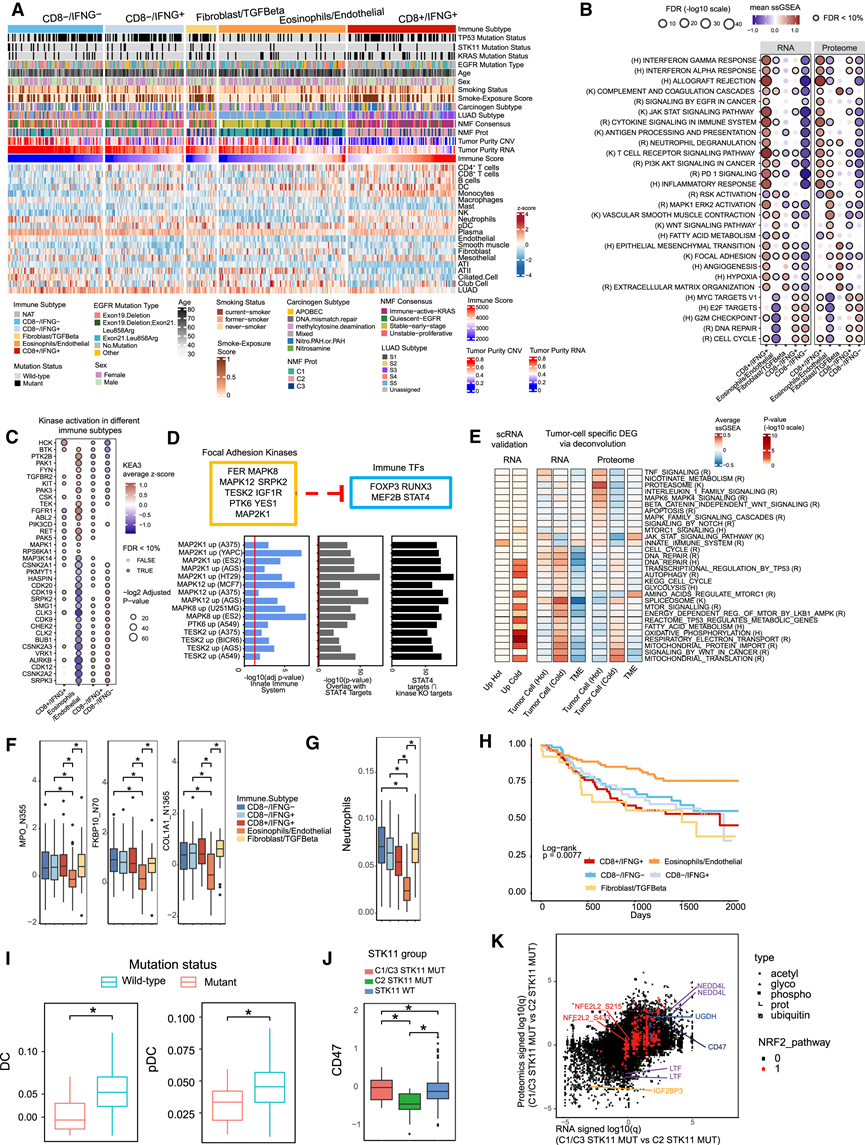
图4. LUAD 的免疫概况。
(A) 注释轨迹显示从蛋白质组数据得出的免疫亚型、通过 RNA-seq 数据的 ESTIMATE 得出的免疫评分、通过 RNA-seq 数据得出的肿瘤纯度、从 CNV 数据估计的肿瘤纯度以及各种临床和突变参数。(B) 气泡图显示了免疫亚型与生物通路关联分析的汇总统计数据。(C)气泡图显示肿瘤样本免疫亚型与激酶活性评分之间的关联分析。(D) 热肿瘤中与免疫相关 TF 聚类相关的粘着斑相关激酶聚类。(E) 基于RNA(左)和蛋白质组数据(右)的细胞类型特异性通路分析,以表征热肿瘤和冷肿瘤细胞中的通路激活情况。(F) 按免疫亚型分层的选定糖基化位点。(G) 按免疫亚型分层的中性粒细胞浸润。 (H) 按免疫亚型分层的无进展生存期 KM 曲线。(I) STK11突变状态与肿瘤样本中不同细胞类型比例的关联。(J) CD47 按STK11突变状态和 NMF 蛋白质组亚型分层。(K) 散点图显示 C1/C3 STK11突变体与 C2 STK11突变体样本之间的分子畸变。
05
环境致癌物对 LUAD 发展的独特贡献
为了研究主要内源性致癌物和环境暴露对不同地理区域和遗传背景的肺癌的影响,他们根据源自暴露于环境致癌物的人类癌症和多能干细胞的突变特征的相对贡献对肿瘤进行了无监督聚类,揭示了 10 个聚类,这些聚类进一步聚类为四个主要组和一个混合突变特征组(图 5 A)。多环芳烃 (PAH) 和硝基多环芳烃、来自烟草或空气污染的致癌物在吸烟者和高烟雾暴露评分从不吸烟者的肿瘤中占主导地位,与地域无关。这些肿瘤富集 CD8-/IFNG-(在EGFR/KRAS -WT 中)或 CD8+/IFNG+(在KRAS突变中)免疫亚型(图 5B)。相比之下,高亚硝胺特征反映了加工食品、化妆品和香烟烟雾中发现的强致癌物,主要发生在患有早期EGFR-L858R或 EGFR- E19Del突变肿瘤的亚洲女性从不吸烟者中(图 5B)。PAH/硝基 PAH在吸烟者和患有KRAS突变或EGFR -WT 肿瘤的从不吸烟者中富集,而亚硝胺则特定于患有EGFR突变的从不吸烟者(图5C)。低亚硝胺肿瘤包括一小部分具有独特 APOBEC 特征的亚群(“突变组”分配中的黄色)(图 5 A-B)。四大组在无复发生存期 (RFS) 和 OS 方面存在显著差异(图 5 D)。PAH/硝基 PAH和 APOBEC 组肿瘤患者的 RFS 明显较差,复发事件的风险比(HR)分别为 2.07和 1.87(图 5 D-E)。此外,富含亚硝胺标记的肿瘤(EGFR - E19Del基因富集)的 RFS 和 OS 较差(图 5 F),其RFS的校正HR为6.07。这些发现强调了亚硝胺和硝基多环芳烃在吸烟者和不吸烟者中导致肺癌不良临床结局的关键作用。
烟草烟雾与雾霾污染是PAH、杂环化合物及N-亚硝胺等致癌物的重要来源。为探究其长期暴露如何驱动正常组织癌变,他们分析了携带亚硝胺和硝基-PAH/PAH特征肿瘤的配对NAT中差异蛋白与通路(图5G)。不同致癌物特征组间存在显著重叠,尤其体现在中性粒细胞脱颗粒、MHC-II抗原呈递和FcγR介导的吞噬作用等免疫相关通路上。高亚硝胺特征肿瘤的NAT呈现代谢与免疫激活(AGER信号、CXCR4信号)以及GNRH和ERK-MAPK信号通路活化,可能促进增殖与侵袭;硝基-PAH/PAH相关NAT则显示IL-8信号通路上调。这些结果将致癌物暴露与LUAD癌旁组织的炎症反应、肿瘤发生及进展相关联,并揭示致癌物特异性改变(如亚硝胺诱导的代谢失调)。
吸烟者是外源性致癌物特征富集的主要人群。为区分吸烟相关癌变与非吸烟者的其他致癌物暴露效应,他们采用S评分法比较高低吸烟暴露评分(HSS/LSS)。在226个上调和157个下调特征中,197个整合进蛋白质互作网络并形成19个互连模块。免疫受体、补体系统组分和趋化因子的上调表明HSS NAT微环境中存在免疫应答与免疫细胞募集的复杂调控。通路分析一致显示免疫相关过程及葡萄糖、胆固醇和脂质代谢的显著富集(图5H)。值得注意的是,虽然两组均存在炎症和代谢失衡节点,但硝基-PAH暴露在细胞外基质(ECM)降解、炎症和抗原呈递途径中诱导了更多磷酸化事件上调(图5I),提示其致癌作用较PAH更为显著。

图5. 环境致癌物协同诱导 LUAD 发展。
(A) 10 个基于突变的亚聚类分为四大诱变剂/致癌物组,主要基于硝基多环芳烃(深蓝色)、多环芳烃(浅蓝色)、亚硝胺(绿色)、APOBEC(黄色)和甲基胞嘧啶(灰色)特征的相对贡献。(B) 环境致癌物与临床特征和基因突变关联性总结。(C) 汇总图显示肿瘤中不同的内源性诱变剂和外源性致癌物特征(x 轴)及其富集的EGFR和KRAS突变按吸烟状况(y 轴)分离。(D) 四种致癌物组之间的无复发生存率存在显著差异。(E) 使用风险比 (HR) 和 95% 置信区间 (CI) 绘制致癌物组对无复发生存期影响的森林图。(F) KM 图显示,高亚硝胺特征组患者的无复发生存率明显低于低亚硝胺特征组患者。(G) 基于对 PAH/硝基 PAH 和亚硝胺致癌物特征组患者的 NAT 中差异表达蛋白质。(H) 基于蛋白质-蛋白质相互作用网络的高烟雾暴露评分(HSS)NAT 中的信号通路富集,该网络源自 HSS 和低烟雾暴露评分(LSS)NAT 之间差异表达的蛋白质,并分为 19 个强内部连接模块。(I) 伴有HSS的NAT中,与代谢失衡和慢性炎症相关的蛋白质和磷酸化位点上调,主要来自含有PAH/硝基PAH特征的样本。
06
亚硝胺和PAH/硝基PAH致癌物促进不同的癌症相关通路
在完成癌旁组织分析后,他们通过Ingenuity Pathway 分析评估了外源性致癌物对肿瘤发展的贡献,比较了高、低及无致癌物特征组肿瘤中差异富集的蛋白、磷酸化位点及激活通路。环境致癌物分析显示,亚硝胺特征(CS)主要关联女性非吸烟患者(图5B)。具有亚硝胺特征的肿瘤与其配对癌旁组织共有富集通路,但表现出更强的致癌活性,包括线粒体脂肪酸氧化、炎症及免疫应答,这与亚硝胺暴露的早期毒性效应一致。硝基-PAH/PAH特征组肿瘤呈现烟雾暴露激活通路特征,如异生物代谢AHR通路、中性粒细胞脱颗粒、IL-8信号、mTOR信号及活性氧解毒通路,而亚硝胺特征组中MAPK信号和ERBB信号激活更显著(图6A)。这些致癌物特异性调控与代谢激活和癌变密切相关的蛋白表达:例如硝基-PAH/PAH组中AHR下调提示致癌物结合引发其降解,从而促进免疫激活与癌症进展(图6B)。
硝基-PAH中硝基的加入可使其致癌潜力提升10-1000倍,这种差异在肿瘤组织中比癌旁组织更显著(图6B)。硝基-PAH特征肿瘤表现出更强的芳香族化合物代谢、细胞周期和转移相关通路激活,并特异性富集蛋白酶体、核糖体和ATP合酶亚基,以及参与有机氮/芳香族化合物代谢(如香烟致癌物代谢关键酶CYP1B1)、免疫应答[如CD274(PD-L1)、CD79A)]和基质金属蛋白酶(MMP)的蛋白/磷酸化位点(图6B)。
具有硝基-PAH/PAH特征的非吸烟者与吸烟者肿瘤存在共性与特性差异值得关注。部分吸烟者肿瘤(主要为亚洲EGFR突变个体)呈现低吸烟评分(LSS),同时更强APOBEC和甲基胞嘧啶脱氨特征(图5B),显示化学致癌、免疫应答与AMPA受体转运间的交互作用。PM2.5促进EGFR/KRAS突变驱动肿瘤发生,表现为PM2.5处理小鼠模型中巨噬细胞IL-1β和CD274表达升高。他们在无高吸烟评分的年轻非吸烟女性队列中,观察到CD274表达显著升高及RTK/MAPK/PI3K信号、糖基化代谢、IL1信号和中性粒细胞脱颗粒等癌症相关通路的激活。这些分子特征一致提示该人群对空气污染诱发LUAD更具易感性。中性粒细胞脱颗粒促进肺癌进展与转移,而CD274可抑制其细胞毒性,因此具有硝基-PAH特征的非吸烟者或可从免疫治疗中获益。为推进基于致癌物暴露特征的肿瘤分类,他们建立了一组以免疫相关分子为主的标志物组合,用于鉴定对硝基-PAH/PAH最敏感的吸烟/非吸烟亚群(图6C)。
他们对人体组织样本致癌物特征的分析表明,内源性与外源性致癌物与特定驱动突变及吸烟暴露相关。通过低于EC10浓度的硝基-PAH(1-硝基芘,1-NP)和PAH(苯并[a]芘,BaP)处理不同驱动突变肺癌细胞系(A549[KRAS突变]和PC9[EGFR突变]),验证了其致癌效应。蛋白质组与磷酸化蛋白质组分析显示,1-NP和BaP处理上调了细胞周期、DNA修复、EGFR信号和侵袭相关通路(图6D)。值得注意的是,这些致癌物以剂量依赖性方式在不同驱动突变细胞中诱导差异化的癌症进展信号模式(图6E)。在硝基-PAH/PAH特征肿瘤中观察到的AHR下调及CYP1B1、SERPINE1、PTPN11、RB1、ATP5F1A和TNFRSF10B上调(图6B),在1-NP和BaP处理的细胞中同样呈现一致性调控(图6E)。功能上,1-NP和BaP促进A549细胞迁移与侵袭(与富含硝基-PAH/PAH特征的KRAS突变肿瘤不良预后一致),而PC9细胞无此效应(图6F-G)。相反,1-NP增强PC9增殖但对A549细胞无显著影响(图6H)。这些发现表明,相同致癌物会因基础驱动突变不同而引发差异化癌症表型,凸显环境致癌物作用的复杂性。将致癌物暴露作为综合风险指标进行常规检测,可为化学预防和监测策略提供依据。他们提出的候选生物标志物或能为吸烟与非吸烟者提供可操作的干预指导,其血液检测有望建立环境致癌物高风险暴露的无创评估体系(图6C)。

图6. 亚硝胺和硝基多环芳烃/多环芳烃致癌物促进不同的癌症相关通路。
(A) 基于对 PAH/硝基 PAH 和亚硝胺致癌物特征组患者肿瘤中差异表达蛋白质。(B) 差异蛋白表达谱显示了特定致癌物对 LUAD 肿瘤的影响。(C) 自述吸烟者或从不吸烟者中,HSS 或 LSS 肿瘤中差异性上调的候选蛋白质生物标志物。(D) 对经1-NP(5 μM)或BaP(5 μM)处理的A549和PC9细胞进行Metacore通路差异蛋白质组学和磷酸化蛋白质组学分析。(E)从经 1-NP 和 BaP 处理的 A549 和 PC9 细胞的差异表达谱中筛选出的蛋白质或磷酸化位点,其中轨迹显示 Metacore 通路分析显示其在代谢、EGFR 信号转导、细胞周期和增殖、转移或 EMT 等功能方面富集。(F) 用致癌物 1-NP 和 BaP 处理的肺癌细胞的迁移能力。(G) 致癌物1-NP和BaP处理后肺癌细胞的侵袭能力。(H) 致癌物1-NP和BaP处理肺癌细胞后进行细胞增殖检测。
07
蛋白质组聚类鉴定早期晚期 LUAD 亚型
基于蛋白质表达的NMF聚类产生三个亚群(C1、C2、C3),其中C2在I-III期患者中显示最短中位无复发生存期(RFS,2.8年),而C3预后最佳(图7A)。C2包含高比例晚期样本(III期58%,IV期50%),复发率是其他组的1.7-2.1倍(60%患者复发),癌症相关死亡最多(52%),淋巴结转移率更高(pN2占58%)且ECOG评分更高。该组还包括36.4%的I期患者,即前期研究中定义的"类晚期"亚型。使用前期ICPC.A研究标注的"类晚期"肿瘤训练的极端梯度提升决策树模型预测显示,该类肿瘤高度富集于C2组。与类晚期表型一致,C2组I期患者的RFS在所有I期中最差,且与吸烟状态无关(图7A)。此外,C2组中高吸烟暴露评分的自报吸烟者生存期显著差于其他吸烟者,而非吸烟者的RFS不受吸烟暴露评分影响。通路分析显示,C2亚型样本具有病理晚期(III/IV期)的分子特征,与其临床分期无关(图7B)。相较于其他组,C2样本富集免疫与转移相关通路,如T细胞受体信号、微自噬、Notch信号、TNFR2非经典NF-kB通路及细胞周期检查点(图7B),其中C2非吸烟者较非C2非吸烟者显著上调免疫相关通路(图7C)。他们将I-IV期表达量持续升高且标注为可成药靶点或血液分泌蛋白的分子列为C2患者潜在标志物(图7D)。差异最显著的蛋白中,COL11A1和THBS2已证实通过多机制促进LUAD侵袭性。此外,C2非吸烟者显著过表达与LUAD进展相关的KRT6A和KRT14蛋白。这些结果共同鉴定出一类具有晚期分子特征的早期患者亚群,其预后较差,或需加强监测与治疗干预。
鉴于C2肿瘤的侵袭性特征,他们进一步探究其与非C2样本相比是否存在共性或独特的致癌因素。TP53突变在C2肿瘤中尤为普遍(66.3%),显著高于C1(32.6%)和C3(31.8%)。致癌物突变特征呈现异质性,在三个蛋白质组聚类中表现出群组和性别特异性分布(图7E)。硝基-PAH特征在男性(尤其是C2组男性,71.6%)中发生率显著高于女性。APOBEC特征与性别相关,APOBEC(C>T)在男性(67-80%)中占优势,而APOBEC(C>G)在女性(39-40%)中更常见。值得注意的是,C2蛋白质组亚型包含两种截然不同的临床特征:一类为吸烟男性主导,多为EGFR野生型(70%)伴或不伴KRAS突变;另一类为非吸烟女性主导,大多为EGFR突变(68%),这促使他们分析C2亚型内性别相关的突变特征贡献(图7F)。亚硝胺和APOBEC(C>G)特征主要见于EGFR突变(L858R和E19Del)的C2女性患者,而硝基-PAH和APOBEC(C>T)特征则多见于男性患者,其中PAH特征在高吸烟评分的KRAS突变男性中富集。与非C2亚型相比,C2组还显示APOBEC3A、APOBEC3B和APOBEC3G蛋白上调。
虽然越来越多证据表明APOBEC诱导的遗传异质性促进癌变并成为癌症标志,但其性别差异关联鲜有报道。为阐明C2亚型中EGFR突变富集的非吸烟女性与EGFR WT/KRAS突变富集的吸烟男性肿瘤的分子特征,他们通过通路分析其通路生物学特征。与C1/C3相比,C2样本共同富集癌症进展、细胞死亡与存活、代谢、免疫应答和转移相关通路(图7G),这与早期病理阶段即出现不良预后一致。除C2患者具有更高免疫评分(ESTIMATE)外,还观察到性别特异性差异:女性中EGFR和BCR信号轴高度激活,男性中KRAS通路上调(图7G),提示LUAD进展的不同路径最终导致相同的不良结局。蛋白酶体组分在C2患者中普遍激活,加权相关网络分析显示其与APOBEC蛋白(3A、3B、3G)共表达。值得注意的是,免疫蛋白酶体组分PSMB8、PSMB9和PSMB10在C2女性中升高,而男性仅标准蛋白酶体组分上调(图7G)。研究表明PSMB8/9/10高表达与免疫检查点抑制剂疗效正相关,C2女性患者的活跃免疫应答提示其或可从EGFR靶向联合免疫检查点抑制治疗中获益,免疫蛋白酶体选择性抑制剂可作为潜在联合选择。相比之下,针对蛋白酶体抑制和KRAS突变的治疗可能是C2类晚期男性患者的有效策略

图7. 蛋白质组学聚类识别驱动晚期早期 LUAD 的高级分子特征。
(A) I-III 期肿瘤(左)和仅 I 期肿瘤(右)的无复发生存期 KM 图。(B)对各期肿瘤( I-IV 期,左)、C1- C3聚类(包括各期肿瘤)(中)和仅针对 I 期肿瘤的 C1-C3 聚类(右)中差异表达的蛋白质进行的激活典型通路总结。(C) 对激活的典型通路进行总结。(D) 蛋白质组学C2亚型中“晚期样”早期肿瘤中显著富集的前30种候选蛋白质生物标志物。(E) 蛋白质组聚类 C1-3 中发病频率(x 轴)和诱变剂/致癌物暴露特征中值(y 轴)的性别差异。(F) 蛋白质组学C2亚型中性别相关的诱变剂/致癌物特征分布。(G) C2 蛋白质组亚型中共有及性别特异性激活通路富集的总结(左图)。EGFR 和 BCR 信号相关蛋白以及免疫蛋白酶体在 C2 女性中高度激活,而 C2 男性患者的 KRAS 通路和标准蛋白酶体蛋白上调。C2 亚型中共有的激活蛋白参与免疫反应、细胞死亡和存活、代谢和转移(右图)。
08
具有潜在转化效用的候选疗法和生物标志物
他们采用了一种整合肿瘤特征与细胞系功能基因组学的治疗靶点优先排序方法。若某蛋白、激活磷酸化位点或其他PTM位点在特定LUAD亚型(EGFR、RBM10、STK11突变及ALK融合)的肿瘤中较配对癌旁组织显著上调,且shRNA敲低该基因在对应亚型细胞系中导致生存缺陷,则判定为预测药物靶点。该方法筛选出的前15个过表达可靶向依赖性靶点涵盖多种基因组异常,与本研究的其他发现相互印证(图8A)。剪接因子SF3B1在多种基因异常背景下过表达,与既往泛癌分析结果一致。MET鉴定为EGFR-L858R肿瘤的过表达依赖性靶点。MET抑制剂已成功用于EGFR突变合并MET扩增的肺癌患者,证实了该策略识别治疗机会的有效性。核输出蛋白XPO1作为多种肿瘤抑制因子和生长调控蛋白的转运体,在多重分子背景下均显示依赖性。XPO1抑制剂Selinexor已获批用于弥漫大B细胞淋巴瘤,目前正在包括NSCLC(NCT03095612)在内的多项临床试验中进行评估。基于蛋白质组的治疗靶点图谱既包含LUAD特定亚型独有靶点,也涵盖跨亚型共有靶点(图8A)。
根据靶点的可操作性,他们将靶点分为五个层级。第一层级包含11个基因(如EGFR),已有获批的靶向药物;第二层级包含6个基因,其靶向药物已获批用于其他适应症,具有药物重定位潜力;第三层级的10个基因(如热休克蛋白HSP90AA1)有在研或临床前药物,可作为实验性疗法的分层生物标志物;第四层级包含8个小分子抑制剂常见靶点蛋白家族基因;第五层级包含5个膜蛋白,是抗体或CAR-T治疗的潜在候选靶点。这些结果不仅提名了治疗候选靶点,还揭示了相关生物学机制。例如,在EGFR野生型和突变型肿瘤中,多个EGFR泛素化位点均较癌旁组织过表达(图8A),但K716位点泛素化仅在EGFR突变亚型中特异性过表达(尽管EGFR蛋白表达下调)(图8B)。携带EGFR突变的LUAD细胞系对EGFR敲除的依赖性也显著强于野生型细胞系(图8B)。K716泛素化触发活化EGFR的内化,而快速内化的突变型EGFR可能成为抗体偶联药物(ADC)的理想靶点,因为ADC抗原的内化对细胞毒性药物的递送和激活至关重要。类似地,表面受体CSF1R的内化也受泛素化调控,其K812泛素化位点在EGFR-E19del肿瘤中特异性过表达(与蛋白总体水平无关),且EGFR-E19del细胞系对CSF1R表现出选择性依赖(图8C),提示CSF1R可作为EGFR-E19del肿瘤的治疗靶点,而K812位点值得后续机制研究。其他值得深入研究的治疗假说包括在多个LUAD亚型中过表达的热休克蛋白HSP90B1和HSP90AA1(图8A)。PRISM药物筛选显示,STK11突变细胞系对HSP90AA1抑制剂坦螺旋霉素的敏感性高于野生型,而该药物已在肺癌I期临床试验(NCT00004065)中测试,或需按STK11突变状态分层(图8D)。
为蛋白质组NMF聚类样本寻找候选药物靶点,他们首先训练了基于全局蛋白质组的XGBoost分类器预测聚类归属,使用反卷积分析鉴定的100个肿瘤上皮特异性蛋白实现平均AUROC 0.97,随后将其应用于DepMap中的LUAD细胞系蛋白质组数据。预测的细胞系聚类与核心NMF肿瘤的Hallmark ssGSEA评分高度一致。他们鉴定了NMF蛋白质组聚类的多个可靶向依赖性(图8E-G):SF3B1(C1/C2)印证了剪接失调在部分肿瘤中的作用;TOP2A与AURKB的共现提示C2不稳定肿瘤可能存在共依赖性,这类肿瘤还高表达IGF2BP3;与不良预后C2肿瘤相关的KIF11已报道可作为LUAD治疗靶点。这些案例表明本研究的方法能有效筛选值得(临床前)评估的潜在靶点。总之,本研究的系统分析构建了LUAD亚型的可成药蛋白质组图谱,凸显了利用多组学数据发现可适用于多种治疗模式的潜力靶点的转化价值(图8H)。

图8. 具有潜在转化效用的候选药物靶点和生物标志物。
(A) 图表描绘了 LUAD 亚型中确定的前 15 个潜在可用药靶点。(B) EGFR 蛋白(上图)和 EGFR_K716 泛素化位点(中图)在肿瘤中的相对丰度,以及EGFR野生型和EGFR突变型肿瘤中相应的 NAT以及来自 DepMap 的 EGFR shRNA 依赖性评分(下图)。(C) 与 (B) 相同,但标注了 CSF1R 蛋白和 CSF1R_K812 泛素化位点。(D) 箱线图比较了STK11突变型或野生型LUAD细胞系对HSP90AA1抑制剂Tanepsimycin (PRISM)的响应。(E) NMF 蛋白质组聚类 C1 肿瘤的潜在药物靶点。(F) 除 NMF 蛋白质组聚类 C2 肿瘤外,与 (E) 相同。(G) 与 (E) 相同,除 NMF 蛋白质组聚类 C3 肿瘤外。(H) 本研究主要转化发现的总结。
+ + + + + + + + + + +
结 论
本研究对来自 406 名不同地理和人口背景患者的 LUAD 肿瘤及其匹配的正常癌旁组织进行了全面的多组学分析,探讨了尚未深入研究的驱动突变的影响、染色体不稳定性在预后中的作用、免疫信号传导模式、内源性诱变剂和环境致癌物的差异性和性别特异性效应,以及具有“晚期样”特征的早期肿瘤的病理生物学特性。本研究提出了针对基因组高度碎片化的不稳定肿瘤和致癌物暴露的候选蛋白质生物标志物,并展示了 LUAD 亚型特异性治疗脆弱性图谱。这些观察结果和相关数据资源推动了针对这种毁灭性疾病的精准管理策略的目标
+ + + + +



