

 English
English文献解读|Cell(45.5):基于宏基因组的人类肠道微生物组、宿主和饮食暴露组的宏蛋白质组学揭示了健康和炎症性肠病的特征
✦ +
+
论文ID
原名:Metagenome-informed metaproteomics of the human gut microbiome, host, and dietary exposome uncovers signatures of health and inflammatory bowel disease
译名:基于宏基因组的人类肠道微生物组、宿主和饮食暴露组的宏蛋白质组学揭示了健康和炎症性肠病的特征
期刊:Cell
影响因子:45.5
发表时间:2025.01.20
DOI号:10.1016/j.cell.2024.12.016
背 景
人类胃肠道 (GIT) 及其微生物群形成一个代谢活跃的界面,对于营养消化、免疫调节和抵御病原体至关重要。饮食信号显著影响肠粘膜功能、微生物群组成及其相互作用网络。破坏这种平衡,尤其是在具有遗传风险特征的个体中,与各种疾病有关,包括炎症性肠病 (IBD)、乳糜泻、营养不良、心脏代谢疾病、癌症和神经退行性疾病。宿主-微生物组-饮食相互作用在调节人类健康方面发挥着至关重要的作用,但对其直接功能评估仍然具有挑战性。
实验设计
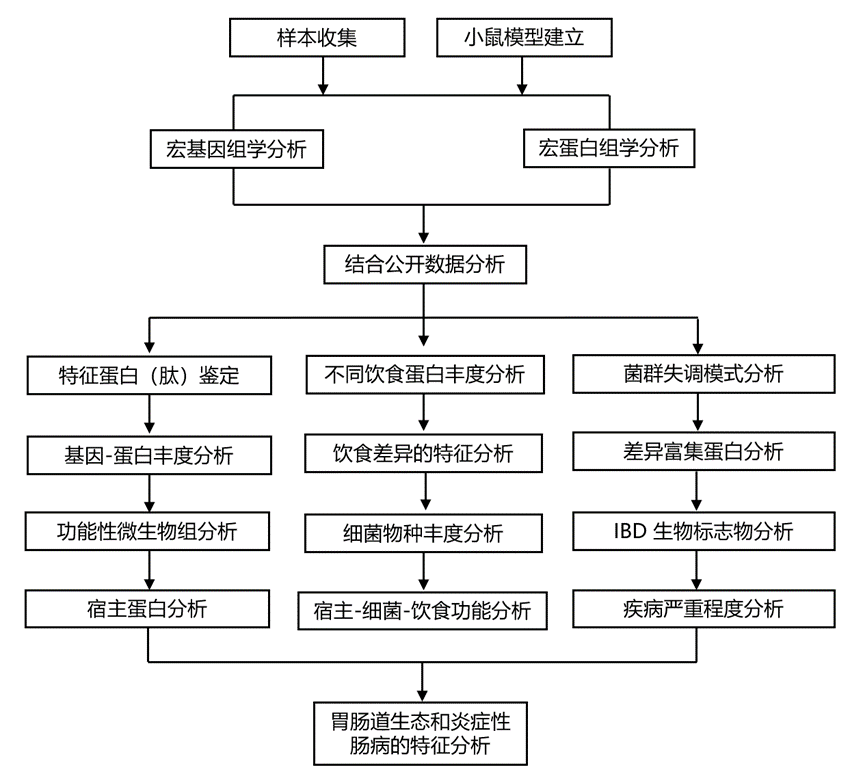
结 果
01

对 MIM 进行调整和基准测试,以同时进行微生物组-宿主评估
研究团队在微生物组中采用了基于宏基因组的宏蛋白质组学 (MIM) 方法,该方法基于每次实验生成的精炼和全面的蛋白质参考目录,可以同时定量来自饮食、宿主及其细菌微生物组的大多数肠道蛋白质(图 1 A-B)。为了方便和清晰,本工作中的结果面板采用颜色编码:宿主蛋白质用蓝色表示,微生物蛋白质用红色表示,膳食蛋白质用黄色表示,微生物组宏基因组数据用紫色表示。MIM 检测到的蛋白质数量与之前的粪便宏蛋白质组学研究相当。为了最大限度地减少由于蛋白质同源性而导致的细菌蛋白质的错误鉴定,并在检测非稀有、以前已表征的细菌物种时获得物种水平的分类学分辨率,他们在构建宏基因组参考数据库时生成了一个逐步过滤过程,该数据库保留了以下内容:(1)相对丰富的物种,代表 95% 的宏基因组信号;(2)序列覆盖率超过 70% 的物种;(3)这些细菌物种可能产生的蛋白质,序列覆盖率超过 75%。总的来说,在健康、正常食物 (NC) 消费、无特定病原体 (SPF) 饲养的野生型 (WT) C57BL/6 小鼠的粪便中,MIM 检测到平均每个样本 631 种宿主蛋白(1955 种独特肽)、2656 种细菌蛋白(5153 种独特肽)和 23 种膳食蛋白(118 种独特肽)(图1C-D)。在健康人类的粪便中,MIM 检测到平均每个样本 363 种宿主蛋白(1,778 种独特肽)、3746 种细菌蛋白(10194 种独特肽)和 58 种膳食蛋白(226 种独特肽)(图 1C-D)。MIM 检测到的微生物蛋白质中只有一小部分源自宏基因组学检测到的真菌、病毒和古菌。
在人类中,与宏基因组分析相比,对健康受试者粪便进行 MIM 分析产生了独特的信号。正如预期的那样,由于某些细菌基因不是组成性表达的,因此宏基因组分析中的 alpha 多样性(即样本内物种丰富度)高于 MIM(图 1 E),后者检测到的蛋白质的基因丰度相对较高。MIM 读数(蛋白质和通路)在样本之间的变化小于相应的基因组读数(基因和通路)(图 1 F),这与观察结果一致,表明与物种水平的群落结构相比,功能性微生物组读数具有较小的个体间变异性。对于大多数检测到的细菌,物种特异性蛋白质的丰度与相应的物种特异性基因的丰度显著不同(图 1 G)。即使在单个细菌水平上(例如Lactobacillus rogosae;图 S1 D),与样本中相应的基因相比,多种蛋白质的丰度也存在显著差异,这可能是由受调控的表达模式驱动的,这表明许多细菌蛋白质无法从基因组数据中推断或“预测”。有趣的是,这种“不可预测”的蛋白质更可能是细胞外的(图1 H),这表明与 MIM 相比,基因组方法在研究分泌蛋白质景观方面可能不是最佳的。
MIM 正确地将 97% 的粪便微生物蛋白质分配到物种水平分辨率(图 1 I)。为了进一步证明利用每个实验的宏基因组参考数据库的优势,他们在精炼的实验人类微生物组蛋白质数据库中补充了不同数量的来自宏基因组学未检测到的物种的随机细菌蛋白质序列。事实上,这种随机添加以剂量依赖性方式显著损害了输出的灵敏度和特异性(图 1 I),并且与宏基因组学未鉴定的细菌物种的蛋白质有关(图 1 J),同时大大延长了计算处理时间至数周(图 1 K)。此外,在复杂的微生物组成中,与使用参考蛋白质目录或基于基因组组装的方法的蛋白质组学分析相比,MIM 显示出检测到的细菌蛋白质的物种水平明显更高的分类学分辨率(图 1 L-M)。
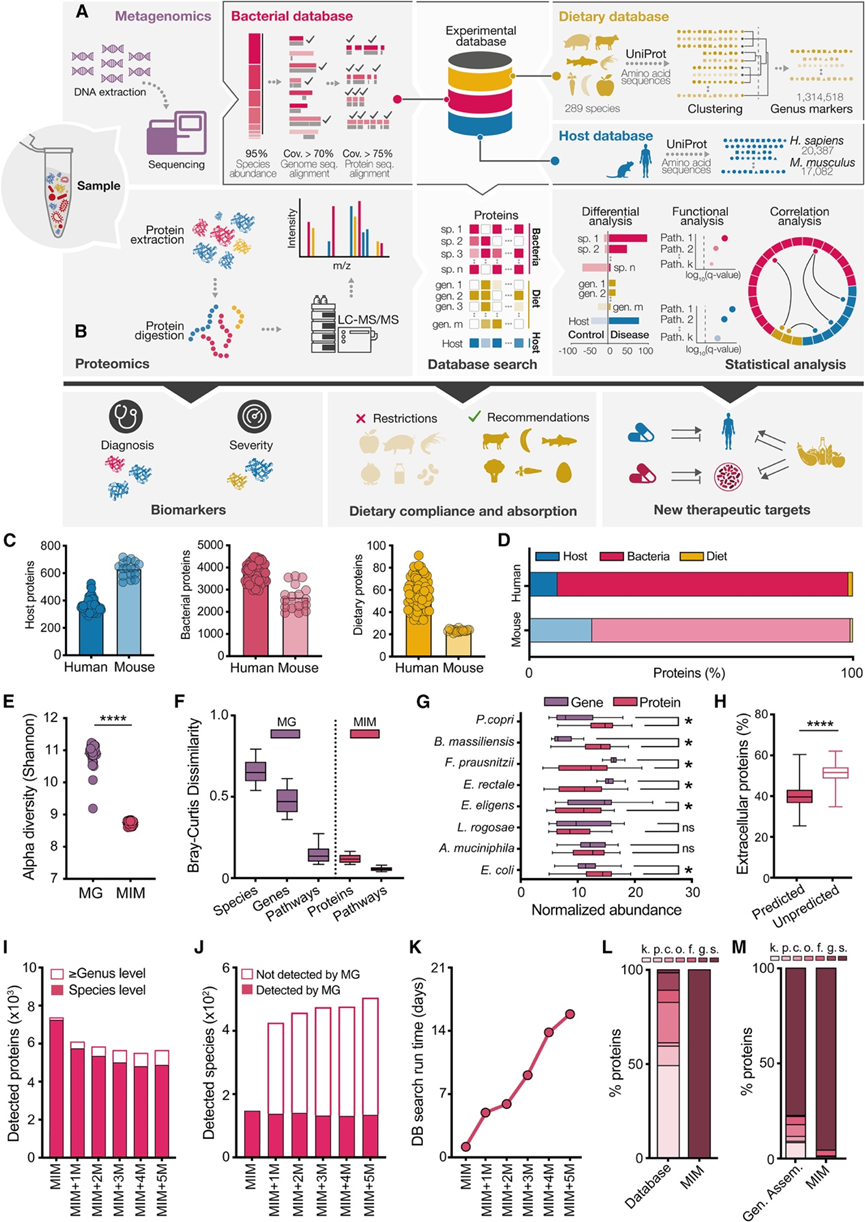
图1. 基于宏基因组的微生物组宏蛋白质组学 (MIM) 分析。
(A-B) 实验示意图。(C-D) 幼稚无特定病原体 (SPF) 小鼠和健康人类中宿主(蓝色)、细菌(红色)和饮食(黄色)粪便蛋白质的总数和百分比。(E)基因和蛋白质水平的 alpha 多样性。(F) Bray-Curtis 差异性。(G) 按细菌种类分层的平均标准化细菌基因和蛋白质丰度。(H) 百分比、预测(相关蛋白质/基因标准化丰度,红色)和未预测(白色)细菌蛋白质。(I-K) 计算机验证 (GER-HC),MIM 蛋白质数据库补充了 1-5 百万个随机、MG 未检测到的细菌蛋白质。(L-M) 蛋白质分类分辨率。
02
MIM 评估人类胃肠道功能性微生物组-宿主相互作用
接下来,他们通过评估一组健康的未接受过治疗的个体和一组接受过广谱抗生素治疗(ABX)的成年志愿者,评估了人类胃肠道中的利基特异性 MIM 信号。所有参与者均接受了上、下内窥镜检查,以获取来自胃、十二指肠、空肠、回肠末端、盲肠、降结肠和粪便的管腔和粘膜样本。在未接受过治疗或抗生素治疗的健康成人中,基于 MIM 的肠道微生物组物种解析组成展示了整个胃肠道中不同的共生活性(图 2 A)。例如,MIM 在一个个体中发现了由幽门螺杆菌产生的 39 种胃蛋白(图 S2 A),该个体表达 Cag 致病岛 (cag PAI) 蛋白,这种蛋白与慢性活动性胃炎、消化性溃疡病和胃癌有关,以及与硝基咪唑耐药性相关的氧不敏感 NAD(P)H 硝基还原酶。在未接受治疗的个体中,检测到的细菌蛋白质的数量和种类沿胃肠道远端增加(图 2 B),这与之前的宏基因组细菌密度梯度发现一致(图S2 B)。基于MIM的生态位特异性蛋白质共生配置在多个特征上与散弹枪宏基因组测序确定的数据不同(图S2C-H)。例如,MIM数据显示 (而不是宏基因组测序数据),唾液链球菌表达的 53 种碳水化合物利用蛋白在胃样本中明显比粪便中丰富得多(图 S2 C-D)。
与健康个体相比,广谱抗生素治疗的特点是具有独特的生态位特异性蛋白质谱(图 2 A),这对于兼性厌氧菌的扩张非常显著,大肠杆菌和肠球菌产生的蛋白质比例在胃肠道中较高,同时伴有蛋白质(图 2 B)和物种(图 S2 B)梯度的损失。在未经治疗的个体中,在下胃肠道中检测到与对多种抗生素耐药性相关的细菌蛋白(图 2 C)。接受抗生素治疗的参与者在物种分辨的抗生素耐药谱中表现出明显的个体化改变。值得注意的是,大多数基于 MIM 的抗生素耐药蛋白 (ARP) 在粪便中检测不到,这表明粪便分析可能无法可靠地确定抗生素耐药性库的存在(图S2C)。
人类胃肠道中的宿主蛋白分布可分为三个主要聚类:(1)胃;(2)十二指肠和空肠;(3)回肠、大肠和粪便(图 2D-E)。在这些聚类中鉴定出多种独特的生态位特异性宿主蛋白。有趣的是,抗生素治疗对这种三聚类宿主结构没有影响(图2D-F)。然而,在不同的胃肠道区域都观察到了受抗生素治疗持续影响的不同宿主蛋白。例如,26S 蛋白酶体的非 ATPase 调节亚基 2 和 3(PSMD2 和 PSMD3)在接受抗生素治疗的个体的下胃肠道中显著减少。泛素-蛋白酶体系统功能障碍与IBD、结直肠癌和胃肠道感染有关。
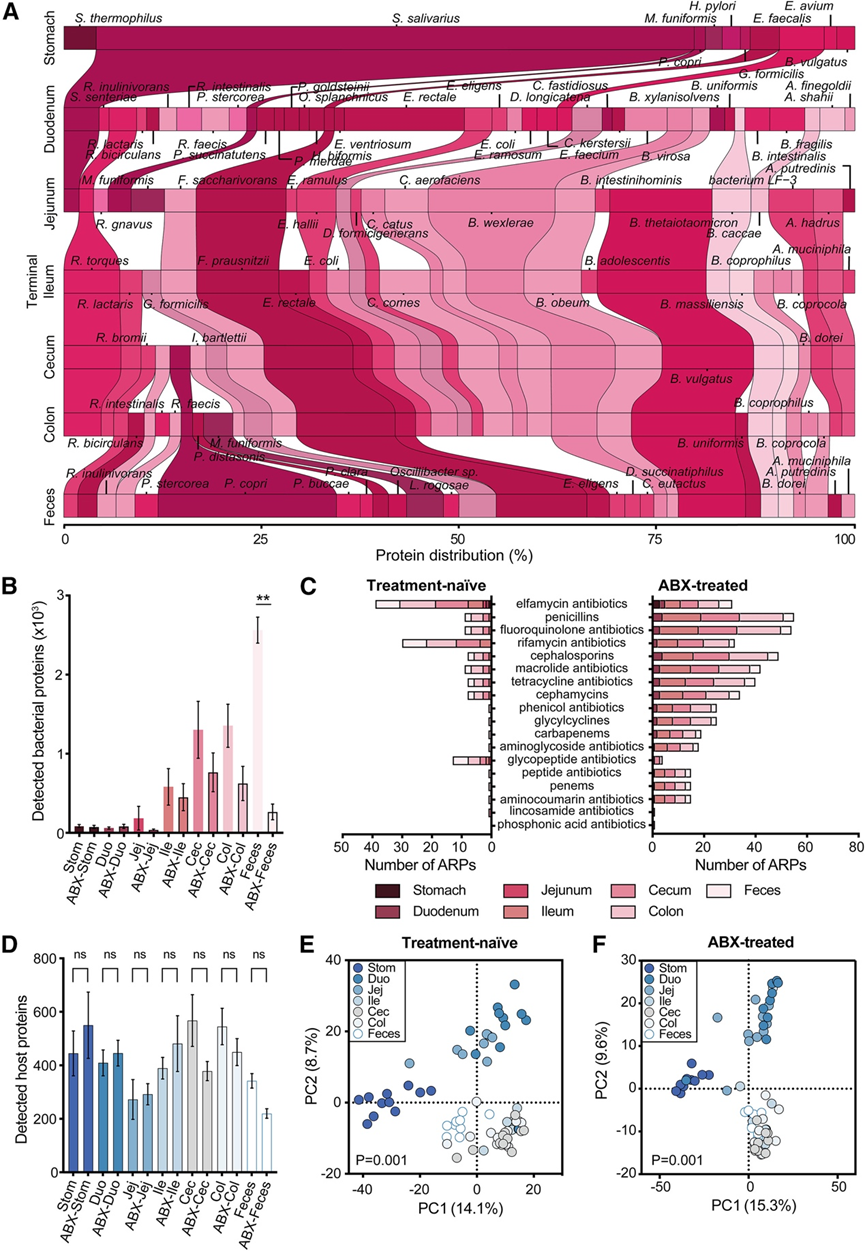
图2. 健康和抗生素干扰人类中的宿主微生物组 MIM 景观。
(A) 细菌蛋白质百分比。(B) 细菌蛋白质总数。(C) 抗生素耐药蛋白(ARP)的总数。(D) 宿主蛋白总数。(E-F) 宿主蛋白总数。
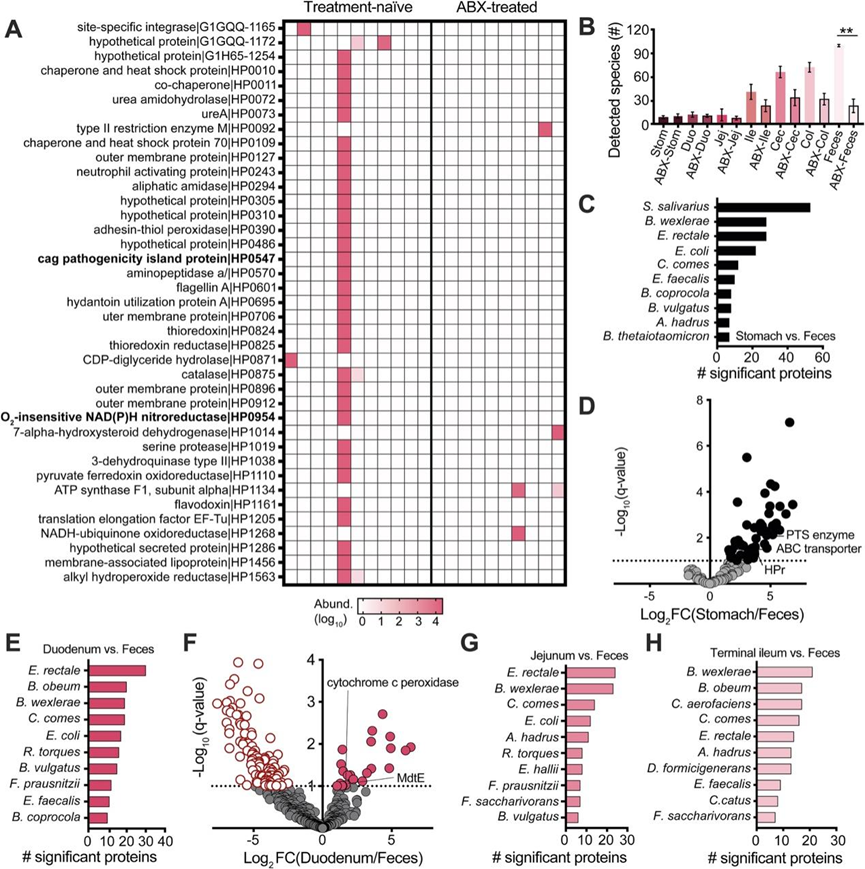
图S2. 抗生素对人类胃肠道微生物组 MIM 谱的影响。
(A-B) 未接受过治疗和接受过抗生素(ABX)治疗的健康成人的粪便和腔内样本。(C) 对于每一种细菌,与粪便相比,胃中重要蛋白质的数量在基因水平上并不显著。(D) 唾液链球菌蛋白质在胃和粪便中的差异丰度。(E) 对于每种细菌,与粪便相比,十二指肠中重要蛋白质的数量在基因水平上并不显著。(F) 十二指肠与粪便中大肠杆菌蛋白质的差异。(G-H) 对于每种细菌,与粪便相比,空肠或回肠中的重要蛋白质数量在基因水平上不显著。
03
饮食暴露的蛋白质组学定量
在小鼠基准实验中,MIM 准确地确定了四种常用啮齿动物饮食中所有食物成分的属丰度:NC 包含来自大豆 (Glycine)、玉米 (Zea) 和小麦 (Triticum) 的蛋白质;HFD(研究饮食 D12492i),包含来自大豆 (Glycine) 和酪蛋白的蛋白质;高蛋白和低蛋白饮食(分别为 Teklad 90016 和 91352),几乎完全由酪蛋白组成。在从 NC 喂养的幼稚 SPF 小鼠获得的粪便中,MIM 检测到 40 种不同的饮食蛋白质,其中至少有 1 种独特的肽(总共 41 种饮食蛋白质)源自小麦、大豆或玉米(图 1 I)。MIM 在接受仅酪蛋白饮食的小鼠中专门检测到了 α-酪蛋白的存在(图 3 A-B),在食用基于氨基酸的要素饮食的小鼠中完全丢失了饮食信号(图 3 A-B),在施用包含这两个属的蛋白质补充剂的小鼠中专门检测到了豌豆 (Pisum) 和大米 (Oryza)(图 3 C),并且准确区分了小鼠的无麸质和含麸质饮食(图 3 D)。重要的是,利用最近引入的估计饮食暴露的宏基因组学流程(基于宏基因组的饮食摄入量估计 [MEDI])显示出低特异性,仅检测到小麦序列。
除了饮食暴露组之外,饮食 MIM 还可以表征消化功能的利基特定变化。这种能力在高脂饮食(HFD)喂养的SPF小鼠的胃腔样本中得到了体现。胃蛋白质消化主要由胃蛋白酶介导,而下游肠道蛋白质分解和随后的体外MIM 蛋白质加工由胰蛋白酶介导。有趣的是,在 MIM 处理的 HFD 喂养小鼠的胃样本中,他们观察到表现出完全胰蛋白酶化特征的肽百分比明显较低(图 3 E),而具有胃蛋白酶-胰蛋白酶特征的肽百分比较高(图 3 E),这表明 HFD 基蛋白质在胃中消化得越来越多,随后迅速吸收。蛋白质覆盖率(检测到的肽所代表的蛋白质序列的百分比)在胃中最高,随后在小肠中降低,在 NC 和 HFD 喂养的小鼠中呈现出不同的模式。
为了评估基于 MIM 的饮食量化在人类环境中的效果,他们首先招募了六名健康志愿者参加一项干预性饮食试验,试验内容包括控制两对植物性食物的消费:香蕉和苹果(分别至少 300 和 400 克)或黄瓜和花生(分别至少 350 和 100 克)(图 3F)。粪便 MIM 分析通过在各自的消费期后不久专门检测四种饮食元素来准确反映饮食暴露,每种食物使用多种不同的蛋白质(图 3G)。相比之下,将相同样本的粪便宏基因组DNA读取映射到饮食分类数据库或使用基于测序的MEDI流程进行分析,饮食成分的检测能力较低且不具特异性。此外,一项逐步增加花生摄入量的试验(8克、20克和40克花生)表明,粪便中的饮食MIM能够通过检测15种不同的花生蛋白,以灵敏且剂量依赖的方式检测到花生的摄入(图3H)。除了测试食物外,粪便中还能够检测到多种其他摄入的食物项目,反映了参与者在记录的时间段内经历的不同营养暴露。为了进一步评估基于MIM的营养评估敏感性,他们分析了15名健康人群体,该群体是Shalon等人已发布临床研究的一部分,研究报告了在3天跟踪期内所消费的所有食物。实际上,基于MIM的粪便饮食评估在85%以上的情况下识别出了报告中提到的食物项目的蛋白质证据,且在检测任何特定食物时没有表现出敏感性降低。同样,MIM准确捕捉了儿童乳糜泻患者在开始无麸质饮食后发生的饮食变化,同时正确识别出一名未遵循无麸质饮食的儿童。
利用 MIM 对几个人类队列中的整体饮食暴露进行分析,结果显示营养成分存在很大差异,这可能反映了参与者之间不同的地理和文化饮食差异(图 3 J)。值得注意的是,基于 MIM 的营养评估可以准确捕捉由营养消费习惯的不同文化差异所驱动的基于人群的食物暴露差异。例如,基于 MIM 对以色列和德国队列的饮食暴露进行比较,发现两个国家居民粪便中含小麦食物的饮食暴露水平相似(图 3 K)。然而,德国人的粪便中与猪肉相关的 MIM 信号显著增加,而以色列队列中几乎不存在该信号(图 3 L)。这种差异可能是因为犹太人和穆斯林(绝大多数以色列人)由于宗教限制往往严格不食用猪肉制品。重要的是,这种显著的差异源于 14 种具有独特肽的猪肉相关蛋白质在粪便中的含量更高(图3M),凸显了 MIM 对此类饮食暴露模式进行稳健量化的能力。有趣的是,与德国人相比,以色列人对猪肉相关蛋白的暴露缺失“被”更高水平的鸡肉源蛋白暴露所“弥补”(图 3 N),将基于测序的 MEDI 流程应用于相同的德国和以色列数据集未能捕捉到这些营养暴露差异。
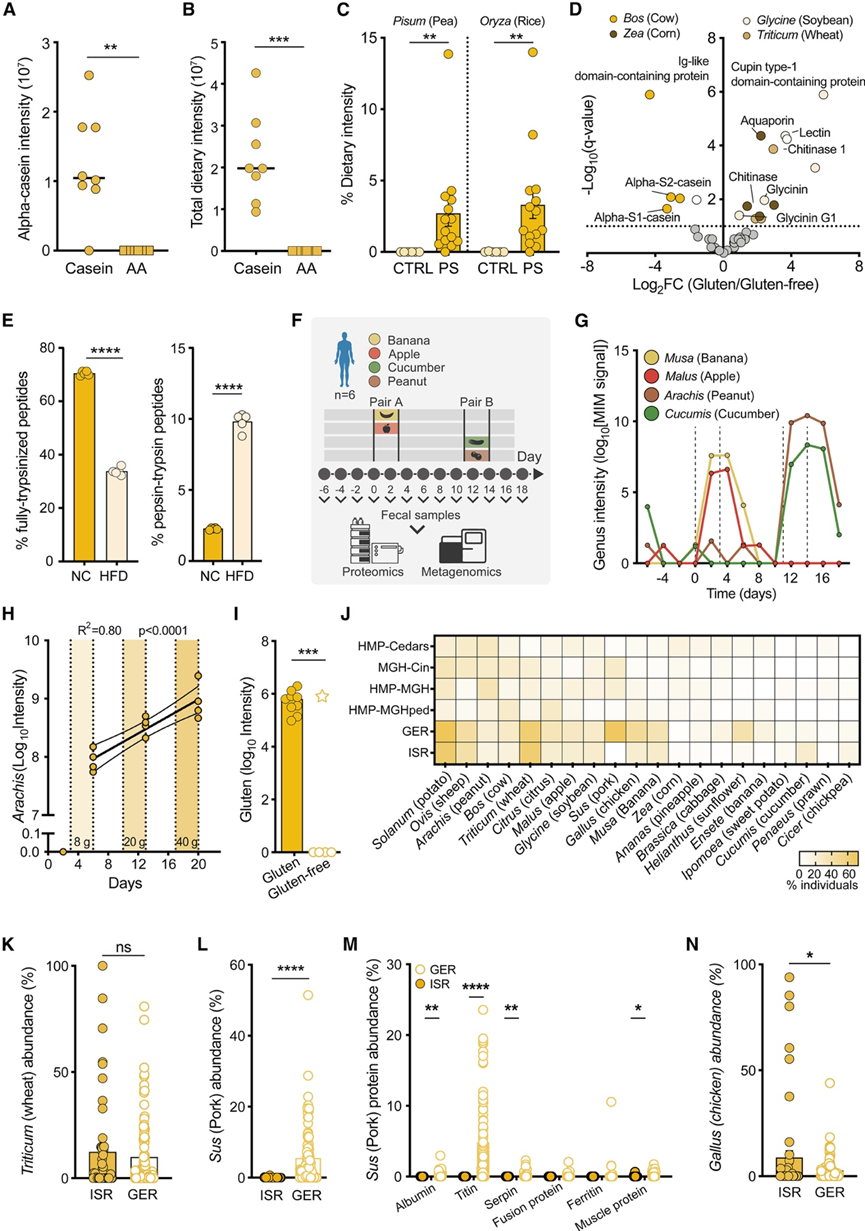
图3. MIM 准确识别小鼠和人类的饮食状态。
(A-B) α-酪蛋白(S1/S2 同工型组合)和总饮食强度。(C) 相对饮食强度。(D) 膳食蛋白质含量差异大。(E) 检测到具有全胰蛋白酶(左) 或胃蛋白酶-胰蛋白酶 (右)谱的胃肽。(F)实验设计。(G) 平均粪便膳食 MIM 信号强度。(H) 粪便花生 MIM 信号。(I)粪便麸质蛋白丰度。(J) 饮食 MIM 信号。(K-N)不同食物中的蛋白丰度。
04
小鼠肠道自身炎症过程中宿主-共生-饮食交叉调节的功能阐明
接下来,他们利用MIM解析在诱导小鼠急性葡聚糖硫酸钠(DSS)炎症性肠病(IBD)结肠炎模型过程中发生的复杂共生体相互作用。早在DSS补充的第3天,整个疾病过程中,粪便微生物组在通过宏基因组或MIM评估时,相较于基线水平,发生了显著变化。重要的是,MIM识别出了在这一急性炎症过程中不同的菌群失调模式:一种“组成性失调”以共生菌丰度的改变为标志,具体表现为绝大多数物种特异性蛋白质的单向改变的蛋白质表达,这些蛋白质可以通过宏基因组学检测(粉红色)或仅通过MIM检测到(红色)(图4A)。例如,Dorea sp. 5-2和Lachnospiraceae 10-1分别出现了92和73种物种特异性细菌蛋白质的增加,而这些物种的分类丰度在宏基因组学分析中未发生改变(图4A)。此外,一种不同的“功能性失调”以某一物种内一组蛋白质的变化为标志,其中大多数“ 管家蛋白”和相关的宏基因组信号保持不变(深红色)。值得注意的是,这种与小鼠肠道自体炎症相关的功能性失调表现为多种细菌蛋白质的减少,主要来自Lachnospiraceae。例如,Eubacterium plexicaudatum产生的62种细菌蛋白质在第3天显著减少,而其他120种检测到的蛋白质保持不变,这与该物种分类丰度未发生改变一致(图4A)。
为了理清肠道炎症中交叉反应宿主和微生物组改变,他们进行了一系列粪便转移,从幼稚和发炎的 SPF 小鼠转移到幼稚和发炎的无菌(GF)小鼠。他们首先测试了无菌宿主炎症对幼稚微生物组的影响。暴露于 3 天无菌 DSS 的供体 GF 小鼠产生了无菌炎症 MIM 肠道反应。随后幼稚微生物组定植到这种无菌发炎的肠道环境中,导致转移后 2 天内出现明显的共生蛋白质组转变,而定植在无炎 GF 肠道中的微生物组则没有这种转变。重要的是,在这种发炎宿主环境中,副拟杆菌和副拟杆菌 YL27等生物产生的多种蛋白质的表达显著下降。将类似的幼稚微生物组转移到 GF 受体小鼠中,并在无菌条件下暴露于 DSS 更长时间(5 天),在微生物组转移后 2 天,诱发了类似的宿主反应和伴随的微生物蛋白质改变。将幼稚共生体更长时间(4 天)暴露于先前无菌的发炎宿主发现了类似的微生物失活(图 4 E),这与 DSS 处理的 SPF 小鼠中观察到的功能性细菌失活特征相似。为了揭示“发炎微生物组”对幼稚宿主肠道引起的早期相互影响,他们通过在饮用水中口服 3 天 DSS 在 SPF 供体小鼠中诱发急性结肠炎。如上所述,在这些炎症供体环境下,P. goldsteinii、P. bacterium YL27和E. plexicaudatum的蛋白质含量降低。将 DSS 治疗的老鼠粪便以及SPF 供体小鼠的粪便微生物转移到GF 小鼠中,在定植后 2 天内引起了粪便宿主 MIM 谱的显著改变(图 4 F),包括抗菌、代谢和炎症宿主特征的显著上调(图 4F)。将暴露于 DSS 较长时间(5 天)的供体小鼠的类似粪便转移到未接受过试验的 GF 小鼠体内,可引起微生物和宿主肠道反应发生类似的代谢和炎症变化。
他们同时评估了急性 DSS 结肠炎期间的 MIM 饮食信号动态。令人惊讶的是,虽然 DSS 结肠炎期间饮食摄入量没有增加,但在肠道炎症过程中,饮食中 MIM 蛋白的相对粪便丰度随着时间的推移而增加(图 4G)。他们推断,这种信号可能源于“亚临床”小肠消化、吸收和/或基于运动的障碍,导致没有增加蛋白质摄入量,但饮食蛋白质粪便中仍积聚。事实上,虽然 DSS 主要诱发明显的结肠炎症和组织损伤,但其对肠上皮细胞 (IEC) 的非特异性影响可能会促使小肠形态和功能发生亚临床改变。一致的是,粪便膳食 MIM 信号的增加与肽 S100A8 和 S100A9 的水平呈正相关,肽 S100A8 和 S100A9 包括炎症生物标志物钙卫蛋白(图4H-I),这表明肠道炎症和粪便膳食蛋白质积累的肠道驱动因素可能相互关联。有趣的是,粪便膳食 MIM 还因明显的营养特异性动态而显著,在炎症过程中不断发展,包括粪便小麦(Triticum)蛋白质丰度的增加,大豆(Glycine)蛋白质丰度的伴随减少,以及玉米相关蛋白质的稳定富集。由于固体鼠饲料中这些营养物质的相对丰度在整个实验过程中保持不变,这些粪便 MIM 改变可能代表肠道炎症过程中小肠消化功能中不同的营养特异性变化,值得未来的研究。
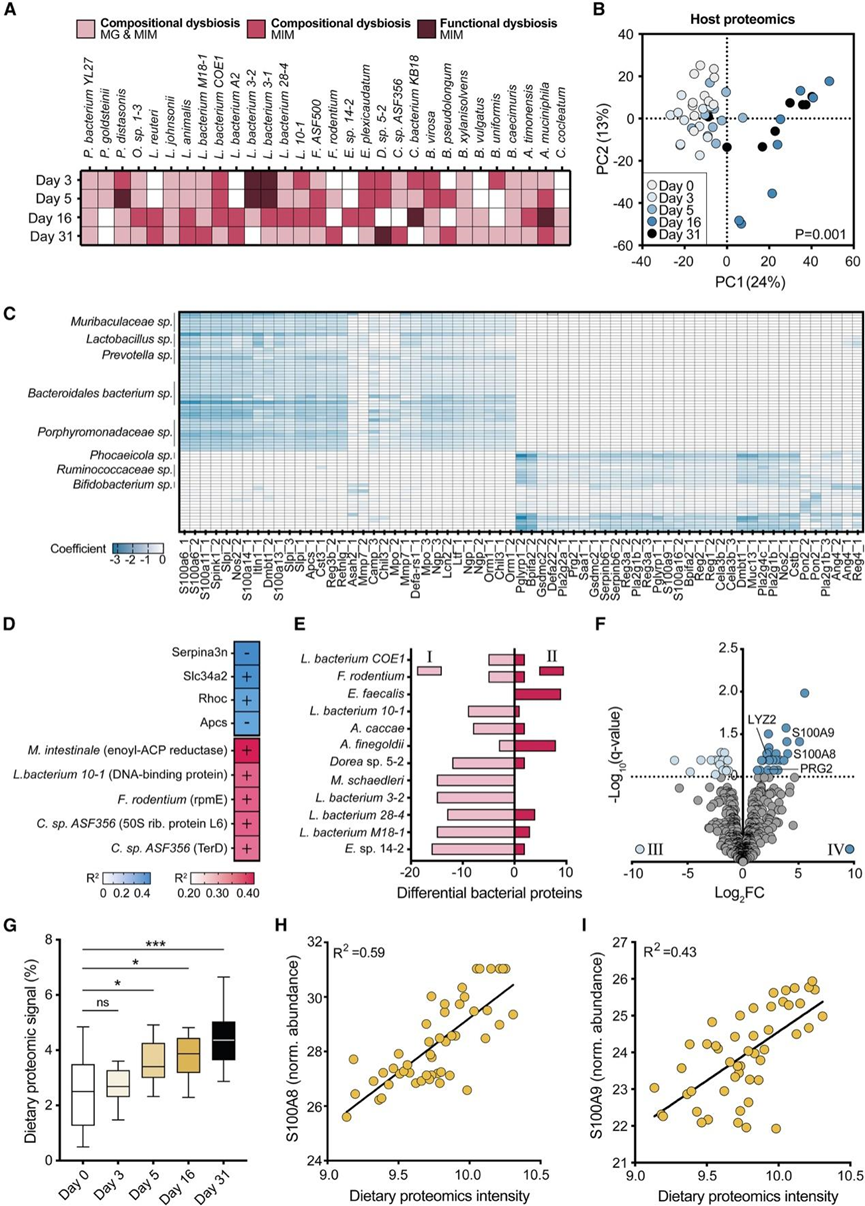
图4. 小鼠肠道炎症过程中宿主微生物组饮食MIM的变化。
(A) 细菌种类。(B) 主成分分析。(C) 宿主抗菌肽与细菌物种丰度相关。(D) 宿主(蓝色)和细菌(红色)蛋白质与疾病严重程度相关。(E) 差异富集的细菌蛋白。(F) 差异富集的宿主蛋白。(G) 在指定时间点检测到的饮食蛋白质组强度百分比。(H-I) 总膳食蛋白质组学强度与 S100A8 和 S100A9水平之间的相关性。
05
MIM 揭示了人类 IBD 中宿主微生物组功能程序的改变
鉴于这些动物模型发现,他们利用 MIM 探索人类 IBD 中的宿主-微生物组相互作用。他们首先评估了一组以色列参与者的粪便 MIM 谱,该队列包括一组新发的儿童克罗恩病患者(ISR-pCD)和年龄和性别匹配的健康对照(ISR-HC)。ISR-pCD 患者粪便中的细菌 MIM 谱与 ISR-HC 有显著不同(图 5 A-B),包括 ISR-pCD 患者中失调的多个物种分辨的细菌代谢途径,例如Bacteroides vulgatus中的聚糖降解和鞘脂代谢(图5B-C)。有趣的是,几种细菌蛋白与临床参数显著相关,包括 C 反应蛋白 (CRP)、钙卫蛋白和儿童克罗恩病活动指数 (PCDAI)。同样,粪便 ISR-pCD 宿主 MIM 特征与 ISR-HC 组明显不同(图5D-E),并且与免疫力有关,特别是中性粒细胞功能和细菌防御(图 5 F)。
与年龄和性别匹配的健康对照者(GER-HC)相比,一组患有溃疡性结肠炎的德国成年人(GER-UC)表现出同样明显的整体微生物 MIM 转变(图5G),包括显著富集的物种分辨途径,例如Segatella copri中的烟酸和烟酰胺代谢(图5H)。与健康对照者相比,宿主 GER-UC MIM 谱也受到整体干扰(图5I),突出显示了与免疫和抗菌反应相关的多种差异丰富蛋白质(图5J-K)。具体而言,AMP 与细菌属之间的相关丰度分析发现多个分类群(Alistipes、Akkermansia和Coprococcus属)与 IEC 产生的 DMBT1 显著相关,并在 ISR-pCD 队列中部分重现。在 ISR-pCD 和成人 GER-UC 队列中,随机森林模型的预测数据表明,与宏基因组学(基于基因丰度)相比,MIM(基于蛋白质丰度)在区分患病对照和健康对照方面具有明显优越的性能(图 5 L)。

图5. 人类 IBD 中的宿主微生物组饮食肠道 MIM 概况。
(A) 主成分分析。(B)KEGG分析。(C) 富集的蛋白质。(D)主成分分析。(E) 差异富集的蛋白质。(F) GO分析。(G) 主成分分析。(H) KEGG分析。(I) 主成分分析。(J) 差异富集宿主蛋白质。(K) GO分析。(L) 诊断分类准确度、受试者工作曲线下面积。
与健康对照组相比,MIM 分析揭示了 IBD 患者的差异富集细菌蛋白数量明显增加,同时发现几乎所有已识别的细菌途径都有更多数量的贡献物种(图 6 A)。同样,在另一组 IBD 患者和 HC中,与基于通用参考数据库的方法相比,MIM 在 IBD 患者和健康对照之间鉴定出了更多的差异富集 蛋白质。
与小鼠一样,MIM 确定了几种与人类疾病相关的菌群失调模式。一种组成性菌群失调模式以S. copri、L. rogosae和Akkermansia muciniphila等物种为特征(图 6B),其大多数蛋白质的丰度变化与其各自的基因相似(宏基因组分类丰度)。第二种组成性菌群失调模式包括Faecalibacterium prausnitzii、Fusicatenibacter saccharivorans和Alistipes putredinis等物种(图 6B),其丰度变化由 MIM 确定,但未由宏基因组学确定。第三种模式由功能性菌群失调组成。例如,青春双歧杆菌(Bifidobacterium adolescentis)和Bacteroides caccae在宏基因组学数据和大多数 MIM 相关的管家蛋白谱中均未发现丰度改变(图 6 B),但某些离散蛋白质的表达发生了改变,例如青春双歧杆菌中的AGE 家族差向异构酶/异构酶和酮醇酸还原异构酶。重要的是,与相应的 HC 组相比,无论是在 GER-UC 成人组(图 6 B)还是在 ISR-pCDIBD 组中,绝大多数此类改变的细菌蛋白质功能均显著下调(图 6 D)。
为了因果探索人类 UC 中功能改变的微生物组对宿主肠道蛋白质分泌反应的影响,他们将健康对照或 GER-UC 患者的粪便微生物组转移到未接受过治疗的 GF 小鼠中。通过宏基因组学或 MIM评估(图 6 E),UC 微生物组的 GF 小鼠接受者的粪便微生物组倾向于与非炎症微生物组的 GF 小鼠接受者的粪便微生物组聚类不同。在 UC 微生物组的小鼠接受者中注意到的组成失调包括宏基因组一致和不一致的 MIM 模式。例如,大多数A. muciniphila蛋白质的丰度降低,类似于它们各自的宏基因组分类学丰度。相比之下,F. prausnitzii中的整体蛋白质减少伴随着未改变的宏基因组分类丰度。除了组成失调外,UC 微生物组的粪便 GF 小鼠接受者中的几种物种还表现出功能性失调。例如,Bacteroides xylanisolvens显示出离散蛋白质表达的改变,整体分类丰度没有差异,包括几种炎症诱导的细菌蛋白的过度表达,例如 Rag/Sus 家族营养吸收外膜蛋白。有趣的是,在 UC 相关微生物组的 GF 小鼠接受者中观察到的与F. prausnitzii、A. muciniphila和Blautia wexlerae相关的功能性菌群失调与GER -UC 供体的情况相似(图 6 B)。
同时,转移了 GER-UC 患者微生物组的小鼠的宿主蛋白质组的聚类趋势与转移了 GER-HC 微生物组的小鼠不同(图 6 F)。有趣的是,在 UC 微生物组的 GF 小鼠接受者中,差异富集的宿主(105 种宿主蛋白)和微生物组(729 种细菌蛋白)蛋白主要与代谢而非免疫功能相关的途径有关。临床上,移植了 GER-UC 相关微生物组的幼稚 GF 小鼠没有表现出结肠炎或全身炎症的特征。这表明,在没有潜在的宿主 IBD 易感因素的情况下,UC 相关微生物组不太可能在幼稚的、基因完整的宿主中急性引发明显的自身炎症反应。
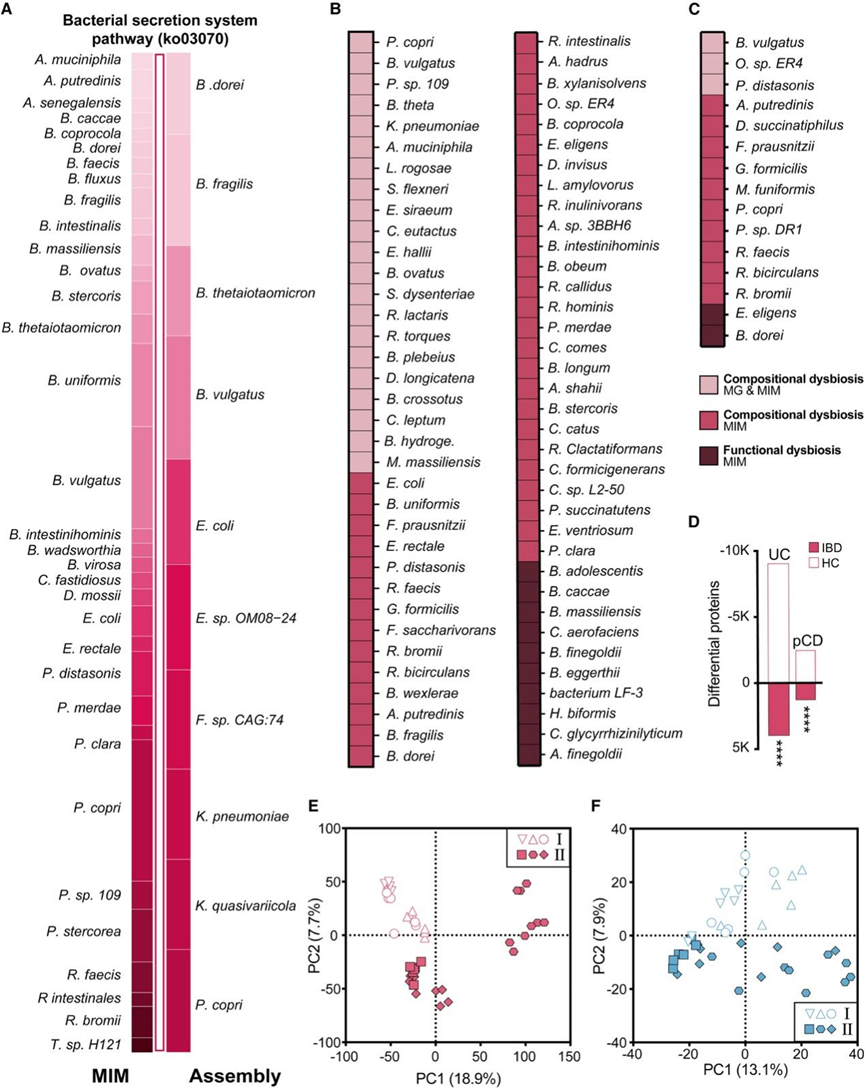
图6. MIM 识别 IBD 中的菌群失调亚型。
(A) 计算机模拟比较,细菌种类、分泌系统途径 KO03070、MIM(左)或基因组组装(右)的相对贡献。(B-C)细菌种类。(D) 按群组划分的健康个体(白色)和炎症性肠病(IBD,红色)中富集的细菌蛋白数量。(E-F)主成分分析。
06
粪便饮食蛋白质组学评估可识别人类 IBD 中的炎症相关蛋白质吸收不良
接下来,他们试图非侵入性地评估营养性 MIM 在人类 IBD 中的可能作用。对新诊断的 ISR-pCD 患者进行粪便营养 MIM 评估,从一线抗炎纯肠内营养 (EEN)饮食干预开始,检测到粪便饮食蛋白信号在 EEN 开始时出现预期的减少(图7A-B),并且具有明显的个体间差异。有趣的是,在这些儿科 CD 患者中检测到的粪便饮食蛋白数量与粪便炎症标志物(即 S100A9)水平之间存在显著的正相关性(图 7C),这表明对 EEN 的依从性和/或相关的肠道炎症减少可以通过不同的 MIM 成分直接和同时量化。
粪便 MIM 营养评估还可以量化人类小肠功能的某些特征,如上图小鼠所示(图 4 G-I)。为了展示这种能力,他们对两个类似处理的 HMP IBD 队列进行了饮食粪便 MIM 分析,辛辛那提医疗中心队列,其特征是上消化道受累的 CD 患者,以及 HMP-MGH 队列,根据内窥镜分类,仅包括下肠道受累的 CD 患者。事实上,在 HMP-Cin 队列中,而非 HMP-MGH 队列中(图 7 D),与健康对照组相比,IBD 患者的总粪便膳食蛋白强度显著增强,这可能表明上肠道小肠吸收不良导致粪便膳食蛋白积累。一致的是,即使在 HMP-Cin 队列中,与内窥镜显示仅有回结肠受累的患者相比,具有上肠道受累的 CD 患者的 MIM 粪便膳食蛋白总丰度也显著增加(图 7 E)。
最后,他们利用 MIM 筛选粪便细菌和宿主蛋白,这些蛋白可以联合用作潜在的 IBD 生物标志物,同时增强和补充临床使用的生物标志物钙卫蛋白(包括 S100A8 和 S100A9)。他们首先利用 HMP 队列中的 HMP-MGH 和 HMP-C,其中 MIM 分别鉴定出 1028 和 770 种粪便疾病相关宿主蛋白以及 13491 和 10631 种疾病相关细菌蛋白。在两个队列中的每一个中,训练交叉验证随机森林分类器在诊断个体是患有 UC 或 CD 还是健康方面显示出高准确度(曲线下面积 [AUC] 超过 0.9)。有趣的是,IBD 预测人类宿主蛋白在 CD 和 UC 之间有明显的重叠。相反,IBD 预测细菌微生物组蛋白大多对 CD 或 UC 具有特异性。在下一个迭代步骤中,他们将 MIM 在上述每个分析中检测到的前 50 个潜在 IBD 判别特征编译成多个单或双蛋白质组合。然后将它们应用于三个验证队列中的每一个的数据:GER-UC 队列、ISR-pCD 队列(ISR-pCD)和 HMP Cedars-Sinai 医疗中心队列(HMP-Ced)。在对每个队列进行独立分析时,通过重复 5 倍交叉验证随机森林模型评估一种或两种鉴别蛋白的随机组合在 UC/CD 诊断中的综合预测性能,并与钙卫蛋白进行比较。在 CD和 UC中(图7F-G),他们鉴定了几对细菌和宿主来源的蛋白质,它们在准确诊断疾病方面优于钙卫蛋白。
为了测试 MIM 预测疾病严重程度而不是疾病存在与否的能力,他们使用了 Mills 等人先前发表的 IBD 队列数据。其中包括基于结肠镜检查的严重程度评分(UC 和 CD 内镜严重程度指数;UCEIS 和 CDEIS)。使用支持向量回归分析,多个 MIM 检测的蛋白质对可以适度预测 CD和 UC中的疾病严重程度水平(图 7I-J),准确度高于钙卫蛋白。一些利用的微生物组 KO 生物标志物,例如 K00133(天冬氨酸半醛脱氢酶),由不同队列中的不同物种贡献(图 7 K),证明微生物组功能可能比微生物组成更能代表健康和疾病状态之间的“共同点”区分因素。在一项试点试验中,通过 ELISA 验证了两种候选生物标志物,该试验包括五名对 EEN 治疗有临床反应的 ISR-pCD 患者的纵向样本(pCD 活性指数 [PCDAI] 和钙卫蛋白。正如 MIM 所建议的,髓过氧化物酶 (MPO) 和髓母细胞蛋白 (PRTN3) 均在临床改善后降低。进一步验证 MIM IBD 标记,再加上生成和优化粪便兼容的基于 ELISA 的检测方法,值得未来的临床试验。
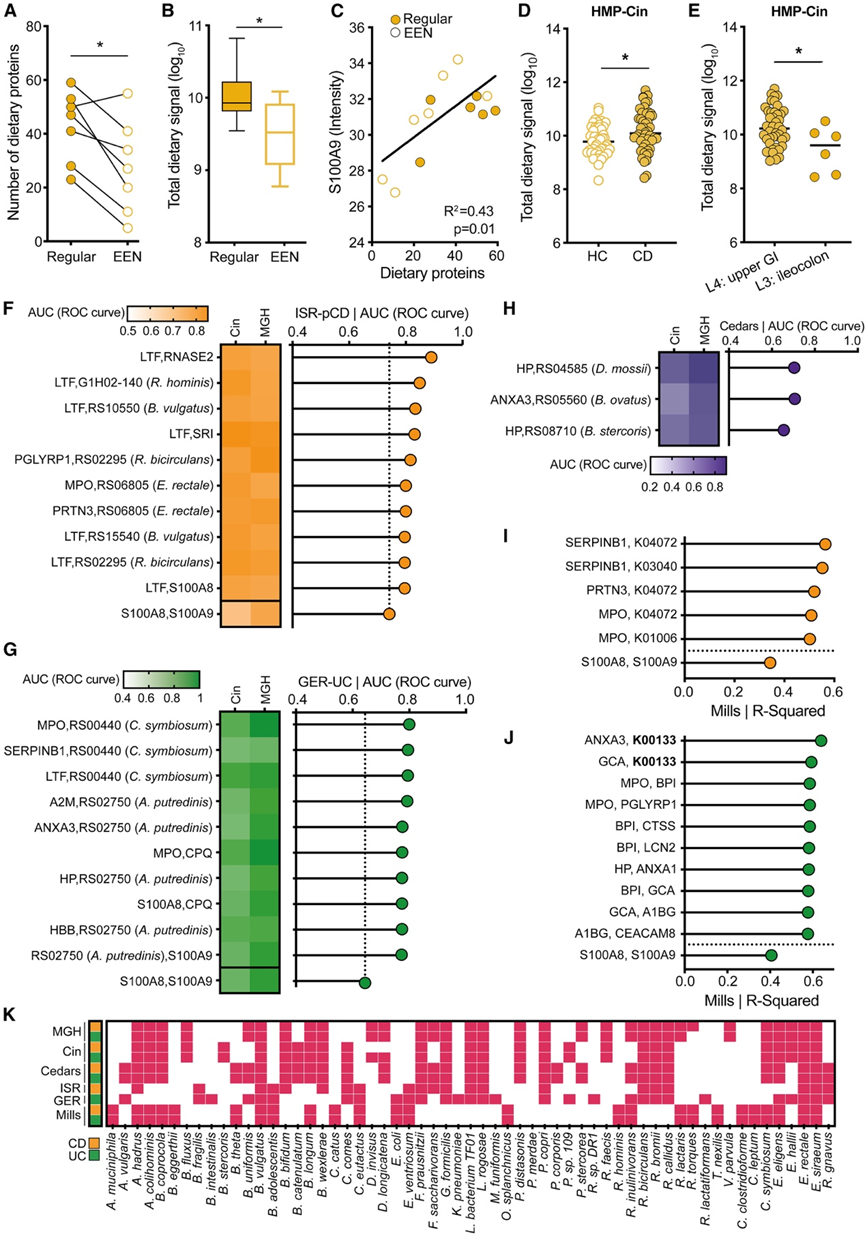
图7. 基于MIM的非侵入性炎症性肠病生物标志物。
(A) MIM 饮食蛋白质的数量。(B) 总 MIM 饮食信号。(C) 饮食 MIM 信号与钙卫蛋白之间的相关性。(D) 总 MIM 饮食蛋白质丰度。(E) 总饮食蛋白质丰度。(F-G) 种蛋白质组合的CD 和UC 诊断的随机森林分类器。(H) 使用宿主和细菌蛋白、HMP-MGH 和 HMP-Cin 队列区分 UC 和 CD 的随机森林分类器。(I-J) 用于 CD 或 UC 诊断的前 50 种判别蛋白随机组合成一到两种蛋白组。(K) 与 K00133(天冬氨酸半醛脱氢酶)相关的细菌种类。
+ + + + + + + + + + +
结 论
本研究在小鼠和人类样本中采用了基于宏基因组的宏蛋白质组学技术,以非侵入性的方式探索共生体和病原体定植、营养改变和抗生素诱导的扰动过程中物种水平的微生物组-宿主相互作用。同时,粪便 MIM 准确地描述了多种临床和饮食环境中的营养暴露状况。在小鼠自身炎症和IBD中进行 MIM分析,结果显示“组成性菌群失调”和伴随的物种特异性“功能性菌群失调”特征,这是由抑制共生体对炎症宿主信号的反应所驱动的。微生物组转移揭示了这些宿主-共生体交叉反应模式的早期发病动力学,而预测分析确定了粪便宿主-微生物组 IBD 生物标志物蛋白对,其表现优于 S100A8/S100A9(钙卫蛋白)。重要的是,同时进行的粪便营养 MIM 评估能够确定 IBD 相关的消费模式、饮食治疗依从性和小肠消化异常。总的来说,并行的饮食-细菌-宿主 MIM 评估在功能上揭示了跨界相互作用组,这些相互作用组塑造了胃肠道生态,同时为微生物组相关疾病提供了个性化的诊断和治疗见解。
+ + + + +



