

 English
English文献解读|Nat Commun(14.7):蛋白质组学表征揭示了表现为亚实性结节的肺癌的发生和进展
✦ +
+
论文ID
原名:Proteogenomic characterization reveals tumorigenesis and progression of lung cancer manifested as subsolid nodules
译名:蛋白质组学表征揭示了表现为亚实性结节的肺癌的发生和进展
期刊:Nature Communications
影响因子:14.7
发表时间:2025.03.11
DOI号:10.1038/s41467-025-57364-x.
背 景
低剂量计算机断层扫描 (LDCT) 筛查的使用大大提高了以放射学亚实性结节 (SSN)形式出现的早期肺癌的识别率。人们普遍认为,以 SSN形式出现的肺癌代表一种独特的亚型,其特点是与放射学实性肺癌相比,其进展缓慢且预后更好。由于SSN表现出明显的异质性生长趋势,因此肺部 SSN 的管理通常是临床决策中的难题。表现出快速进展趋势的 SSN 需要手术切除,而表现出稳定或缓慢生长的 SSN 则需要定期进行 CT 监测。因此,区分稳定的 SSN 和具有高潜在进展的 SSN 对于促进精确的临床管理至关重要。
实验设计
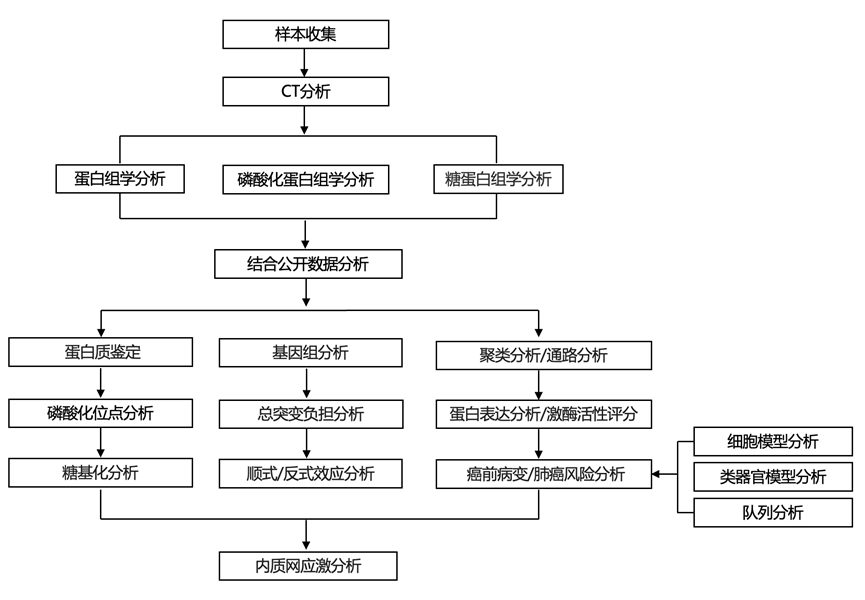
结 果
01

标题
研究团队研究了 66 例放射学显示为 SSN 的肺腺癌 (LUAD) 和 66 例配对癌旁正常组织(NAT)的蛋白质组学景观,这些 SSN涵盖连续的组织学阶段,包括 17 例 原位腺癌 (AIS)、24 例微浸润腺癌 (MIA)和 25 例浸润性腺癌 (IAC),遵循国际肺癌研究协会 (IASLC) 报告的LUAD 组织病理学分类(图1A-B)。临床评估采用实变与肿瘤比率 (CTR)——CT 扫描中最大实体成分大小与整个肿瘤大小的比率,较低的 CTR 对应于侵袭性较低的表型。从 AIS 发展到 MIA 和 IAC,整个肿瘤大小、实体成分大小和 CTR 逐渐增加。验证 LUAD 组织病理学后,对部分冷冻粉碎组织进行蛋白质组学、磷酸化蛋白质组学和糖蛋白质组学表征(图 1B)。
在本研究的队列中,蛋白质组学、磷酸化蛋白质组学和糖蛋白质组学分析共鉴定并定量了10255种蛋白质、27283个磷酸化位点和12480个糖肽。平均而言,每个LUAD样本的蛋白质组约有6700种蛋白质,范围从NAT中的最低约6000种到肿瘤中的最高约7300种(图 1C)。此外,每个样本共鉴定出约7500个磷酸化位点,并具有置信位点定位评分(概率> 0.75)(图 1D),每个样本共鉴定出约3800个糖肽,并具有置信位点定位评分(概率> 0.75)(图 1E)。此外,在所检测的糖蛋白上发现的糖链存在显著的异质性,约 28% 的糖蛋白具有多个糖基位点,47% 的糖基位点发生多个糖基修饰(图 1F-H)。
表皮生长因子受体 (EGFR)、肿瘤蛋白 53 (TP53)、RNA 结合基序蛋白 10 (RMB10) 和粘蛋白 16 (MUC16) 的突变频率分别为 51%、19%、17% 和 17%,与最近对 LUAD 进行的大规模基因组分析的结果一致(图1I)。值得注意的是,亚洲 LUAD人群的 EGFR 突变频率约为 50–60%,而白种人群体中为 15–20%。此外,本研究中的肌联蛋白 (TTN) 突变频率 (29%) 明显高于公开研究数据中的 9%,这可能是因为研究人群中浸润前病变的比例相对较高 (62.1%)。总突变负担 (TMB) 在所有三个组织学阶段均可观察到,并在后期呈现更显著的富集趋势(图 1J)。
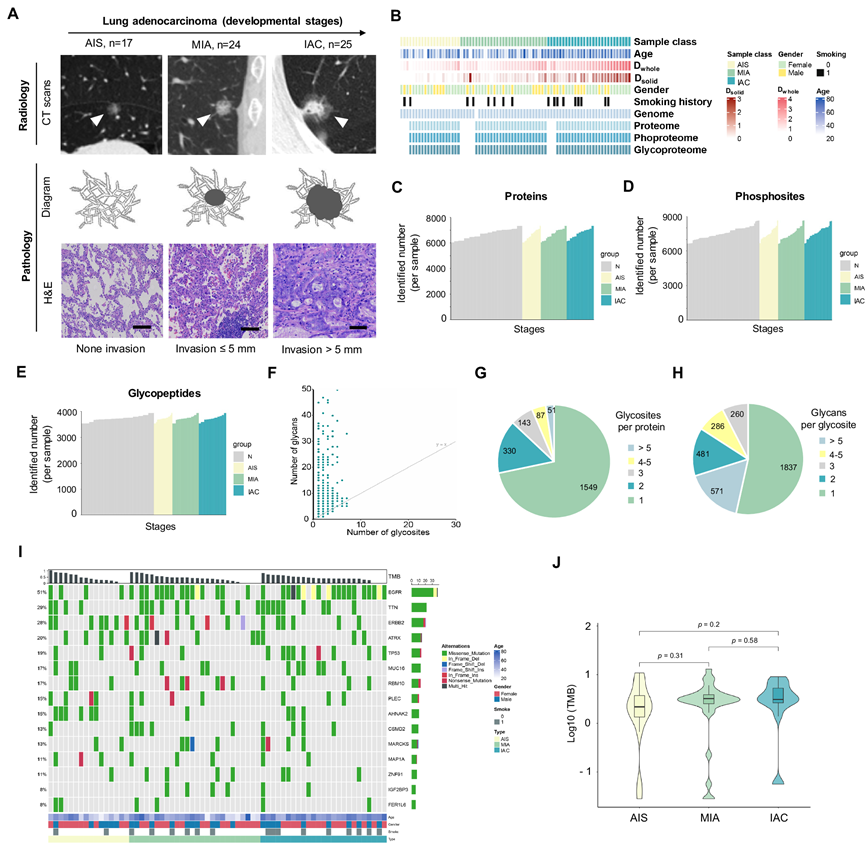
图1. 表现为亚实性结节的肺腺癌的蛋白质组学景观。
(A) 从肺肿瘤前病变到浸润性腺癌 (IAC) 的放射学和组织学发展阶段,表现为亚实性结节。(B) 直方图显示了本研究中的多平台数据和临床特征。(C-E) 肿瘤样本和配对的正常邻近组织 (NAT)中已鉴定的蛋白质、磷酸位点和糖肽的数量。(F) 散点图显示每个糖蛋白中已鉴定的聚糖数量与该蛋白质已鉴定的糖基位点数量的关系。(G) 已鉴定的每个糖蛋白的糖基化位点数量分布。(H) 在给定糖基化位点观察到的不同聚糖数量的分布。(I) 按组织学分期分组的基因组图谱。(J) 突变负担。
02
整合蛋白质组、磷酸化蛋白质组、糖蛋白质组数据
他们检测了影响同源基因产物(顺式效应)或其他基因产物(反式效应)表达的选定突变基因,特别是一组特定的癌症相关基因 (CAG)。有趣的是,他们鉴定出 10 个基因在蛋白质、磷蛋白或糖蛋白数据中具有显著顺式和反式效应。EGFR 突变降低了同源蛋白质,而增加了同源糖蛋白的丰度(图 2A-C)。EGFR 突变肿瘤在 Y62 处显示出 PTPN11/SHP2 的显著酪氨酸磷酸化,但在蛋白质或糖蛋白水平上未观察到影响。有趣的是,他们观察到 EGFR 突变细胞系 (PC-9) 中 SHP2 的 Y62 磷酸化水平高于 EGFR WT 细胞系 (A549、NCI-H226 和 NCI-H1730) (图 2D),表明 EGFR 突变与 SHP2 的 Y62 磷酸化之间存在联系。SHP2 在 Y542 处的磷酸化可以缓解基础抑制并刺激 SHP-2 酪氨酸磷酸酶活性。他们还观察到 SHP2 在 Y542 处的磷酸化模式与 SHP2 在 Y62 处的磷酸化模式相似(图 2D),这与之前的研究一致,该研究表明 SHP2 在 Y62 处的磷酸化可使 SHP2 稳定在开放的活性构象。
据报道,含 sushi 结构域 2 (SUSD2) 的表达存在于多种人类癌症中,例如乳腺癌、卵巢癌和肺癌。在本研究中,EGFR 调节 SUSD2 在 N494 处的糖基化,并且 EGFR 突变肿瘤显示 N494 处的糖基化显著降低(图 2C)。进一步的分析表明,EGFR 突变与从 AIS 到侵袭性 LUAD 的转变中 PTPN11 磷酸化的增加和 SUSD2 糖基化的降低相关(图 2E)。本项研究结果为未来研究糖基化和磷酸化模式作为阻止从 AIS 进展为侵袭性 LUAD 的潜在靶点提供了见解和方向。
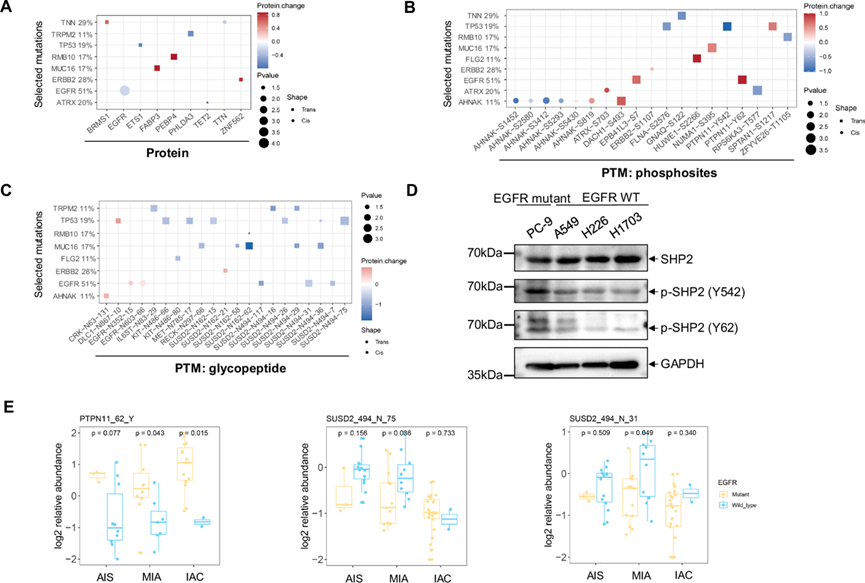
图2.蛋白质组、磷酸化蛋白质组、糖蛋白质组分析。
(A-C) 选定突变(x 轴)对癌症相关蛋白、磷酸化和糖基化表达的顺式和反式影响显著。(D) EGFR 突变细胞系 (PC-9) 和 EGFR 野生型细胞系 (A549、NCI-H226 和 NCI-H1730) 中的 SHP2 磷酸化水平。(E) 箱线图显示在从 AIS 到 IAC 的进展中,EGFR 突变体和野生型 (WT) 样本中 PTPN11 Y62 的磷酸化和 SUSD2 N494 的糖基化。
03
对表现为 SSN 的 LUAD 进行多组学分析,用于治疗干预和早期检测
为了探究蛋白质组学数据的内在结构,他们对蛋白质、磷酸化位点和糖肽进行基于非负矩阵分解 (NMF) 的无监督聚类,并统称为多组学聚类。所有患者在多组学上分为三个聚类。27 名患者分为聚类 1,18 名患者归为聚类 2,21 名患者归为聚类 3,各聚类的样本与独特的临床和分子特征显著相关(图 3A)。聚类 1 与 ECM 受体相互作用、白细胞跨内皮迁移、肾素-血管紧张素系统和细胞粘附分子相关,在女性患者和较小的肿瘤直径中富集。聚类 2 与核糖体、剪接体、溶酶体和蛋白质消化和吸收相关。聚类 3 与细胞凋亡、自噬、铁死亡、糖原分解/糖异生、氨基酸代谢和丙酮酸代谢相一致,表现出应激适应性自食过程,在男性患者和较大肿瘤直径中富集。聚类 1 代表侵袭前阶段,与公开研究中确定的 SI 亚型 (AIS/MIA) 相一致。此外,聚类 1 与公开研究中强调高环境和代谢因素的 SI 亚型相对应。此外,本研究中的聚类 1 同样与公开研究中确定的 C4 亚型(终末呼吸单元)相一致,其特征是 EGFR 突变和在中国个体中流行。MARCKS是癌细胞中PIP3水平的调节因子,在多种人类肿瘤中起着抑癌基因的作用。值得注意的是,其突变在聚类1中很少见(图 3A)。
近年来,糖基在癌症中的重要性日益凸显。他们重点关注聚类相关的糖基,选取每个聚类内上调最显著的前250个完整糖肽,以分析各种糖基在肿瘤聚类中的分布情况。他们观察到,在这些聚类中,含有寡甘露糖、高甘露糖和岩藻糖基化糖基的完整糖肽的分布存在统计学上的显著差异(图 3B)。值得注意的是,寡甘露糖糖肽在聚类 2 中占主导地位,高甘露糖糖肽在聚类1 中占主导地位,而岩藻糖基化聚糖在聚类3 中占主导地位(图 3B)。
糖基化模式受糖基转移酶和糖苷酶表达水平的复杂调控。他们随后的研究通过将糖蛋白质组学数据中获得的完整糖肽丰度与蛋白质组学数据中获得的糖基化酶的蛋白质丰度关联起来,深入探究了控制这些糖基化变化的内在机制(图 3C)。根据糖蛋白底物和糖基化酶的表达差异(在至少一个亚型中肿瘤和非肿瘤之间有显著差异)进行筛选,他们建立了194个完整糖肽的丰度与6个亚型特异性糖苷酶(包括GBA、GUSB、GLA、FUCA1、HEXB和HEXA)之间的相关性(图 3C)。正如预期的那样,大多数底物与指定的糖苷酶之间呈负相关。有趣的是,研究发现,表现出寡甘露糖聚糖糖基化的完整糖肽与这些特定的糖苷酶(GBA、GUSB、GLA、FUCA1、HEXB 和 HEXA)呈正相关,这反映了糖苷酶在生产过程中对将其他聚糖转化为寡甘露糖聚糖的促进作用。这些糖苷酶与寡甘露糖聚糖修饰的完整糖肽之间存在显著的正相关性(图 3D)。此外,糖基化酶表达分析揭示了其从聚类 1 到聚类 3 的显著上调趋势(图 3C),这表明靶向和抑制这些酶的活性可能作为 LUAD 的治疗策略。
比较 AIS 和 NAT 中的差异表达蛋白质和磷酸化位点,发现某些磷酸肽的表达模式与其相应的蛋白质水平不同(图 3E),这些改变的磷酸肽代表潜在的治疗靶点(图 3E)。例如,在 AIS 和 NAT 之间,FGFR3 在 S408 位的磷酸化显著增加,但对蛋白质水平没有可检测到的影响(图 3E-F)。此外,与 MIA 和 IAC 相比,AIS 患者的 FGFR3_S408 水平升高(图 3F)。FGFR3 编码一种跨膜受体酪氨酸激酶,在酪氨酸残基上有六个自身磷酸化位点。有趣的是,磷酸肽富集数据集揭示了 S408 处的磷酸化位点,与已知的 FGFR3 自身磷酸化位点不同。为了评估 FGFR3 S408 磷酸化对激活 PI3K-AKT-mTOR 通路的影响,他们在 PC-9 细胞中过表达了野生型 FGFR3 (FGFR3 WT)、磷酸化模拟 FGFR3 突变体 (S408D) 和磷酸化缺陷型 FGFR3 突变体 (S408A)。值得注意的是,FGFR3 WT 和 FGFR3 S408D 均显著增强了 AKT S473 位点的磷酸化,而 FGFR3 S408A 突变对 AKT 的磷酸化具有显性负向影响(图 3G)。一致观察到PI3K和mTOR的磷酸化模式与AKT S473位点磷酸化模式相似(图 3G),表明FGFR3的S408磷酸化位点在激活PI3K-AKT-mTOR通路中发挥作用。此外,FGFR3 WT和FGFR3 S408D的过表达显著增强了PC-9细胞的增殖,而FGFR3 S408A的转染则适度降低了增殖。然而,FGFR3 S408位点突变对PC-9细胞的侵袭性无显著影响。
在探索 LUAD 进展的替代治疗靶点的过程中,他们通过评估磷酸化底物的丰度来标准化激酶活性,鉴定出五种已知与 FDA 批准的抑制剂相关的激酶(ERK1、ERBB2、SYK、JAK1 和 CDK6),与聚类1 相比,它们在聚类 2 和聚类 3 中的活性增强(图 3H)。他们分别鉴定出 69、48 和 46 种 AIS、MIA 和 IAC 中上调的蛋白质。氧磷酶 2 (PON2)、PON3、SLC34A2、粘蛋白 1 (MUC1)、高尔基体膜蛋白 1 (GOLM1)、晶体蛋白 μ (CRYM)是 AIS、MIA 和 IAC 中的上调蛋白质,根据这些研究,所有这些蛋白质之前都与肿瘤发生中的作用有关。
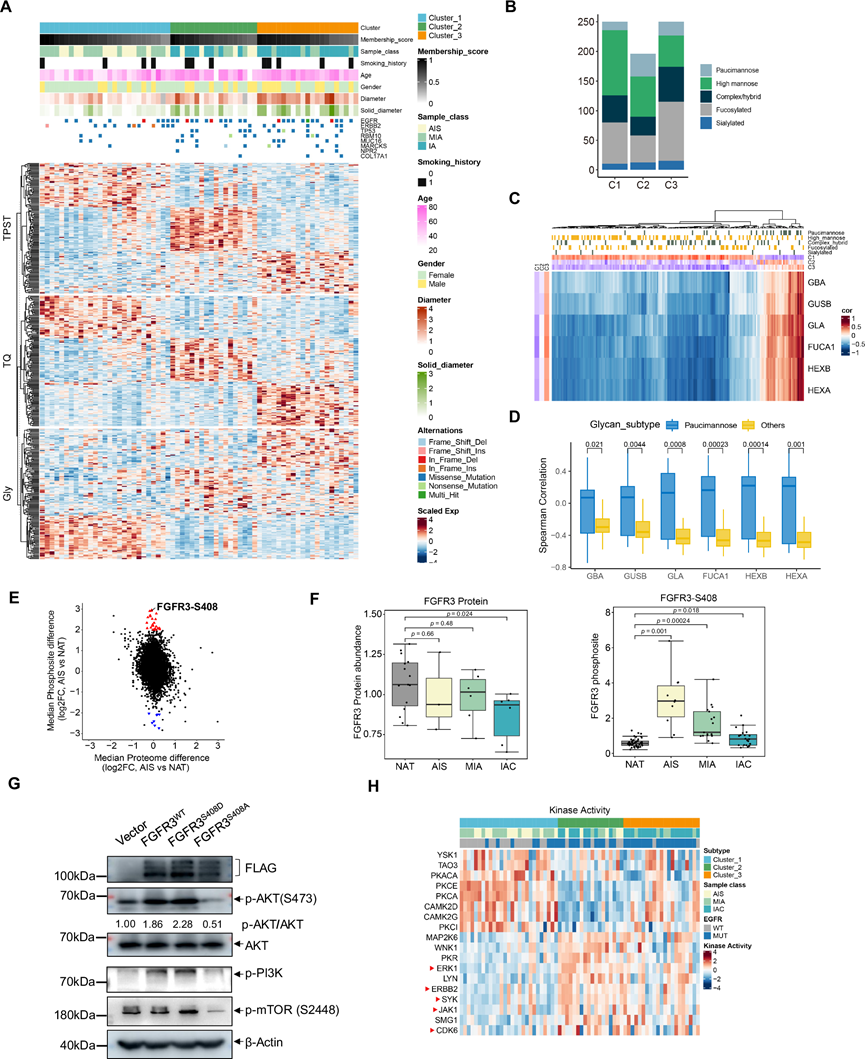
图3. 肺腺癌的多组学分析,用于治疗干预和早期检测。
(A) 将肿瘤综合分类为三个非负矩阵分解衍生的聚类。热图显示每个多组学聚类的前 50 种差异蛋白、磷蛋白和糖基化蛋白。(B) 选择每个聚类中上调最多的 250 种完整糖肽,用于分析肿瘤聚类内的糖分布。(C) 完整糖肽和糖基化酶的层次聚类相关矩阵。(D) 六种选定糖基水解酶与含/不含寡甘露糖聚糖(寡甘露糖等)的完整糖肽之间的相关性。(E) 与正常邻近组织 (NAT) 相比,原位腺癌 (AIS) 肿瘤中磷酸位点差异倍数变化。(F) NAT、AIS、微浸润性腺癌 (MIA) 和浸润性腺癌 (IAC) 中成纤维细胞生长因子受体 3 (FGFR 3) 的整体蛋白表达和磷酸化位点的丰度变化。(G) FGFR3-WT、FGFR3-S408D 和 FGFR3-S408A 对 PC-9 细胞中 AKT、PI3K 和 mTOR 磷酸化水平的影响。(H) 热图显示选定激酶的激酶活性评分。
04
蛋白质组学表征揭示肺肿瘤前病变的发生
他们从蛋白质组学和磷酸化蛋白质组学水平上证实了胆固醇代谢是肿瘤前AIS阶段的一个标志,这可能是驱动LUAD启动的关键分子事件(图 4A-B)。为了验证这一结果,他们选择了胆固醇代谢途径中的重要靶点-前蛋白转化酶枯草杆菌蛋白酶/kexin 9型(PCSK9),软琼脂实验表明,抑制PCSK9可促进永生化人包皮成纤维细胞系HCA2-TERT细胞转化为肿瘤细胞,表明胆固醇代谢在肿瘤发生中起着关键作用(图 4C-F)。PCSK9持续促进LDL受体的降解(图 4D),从而减少LDL从循环中的清除。
为了进一步阐明 PCSK9 在肿瘤发生中的作用,他们在腺病毒-Cre 存在的情况下使用 Kras-LSL-G12D 小鼠的肺组织生成小鼠肺类器官,并分析在有或没有 PCSK9 抑制剂 (Tafolecimab)的情况下类器官中的肿瘤形成情况(图 4G)。这些 Kras-LSL-G12D 肺类器官在培养 7 天后形成上皮球体(图 4H)。随后,采用免疫组织化学 (IHC) 评估肺表面活性蛋白 (SFTPC) 的表达,SFTPC 是肺泡 II 型细胞 (AT2) 的已知标志物。肺组织类器官中 SFTPC 的高表达(图 4I)证实了 AT2 细胞的优势,AT2 细胞是 LUAD 的起源细胞。扩增后,将类器官均等分,并用腺病毒-Cre感染(图 4J)。值得注意的是,与对照组相比,PCSK9抑制剂组观察到更多的类器官(图 4K),并且前者显示出更大的尺寸(图 4L),表明PCSK9抑制剂促进了类器官在体外的生长。通过H&E染色分析类器官的组织学特征(图 4M)。有趣的是,PCSK9的抑制显著降低了SFTPC的表达水平并提高了上皮细胞粘附分子(EpCAM)的表达水平,这表明上皮性肿瘤的形成(图 4N-O)。此外,PCSK9 抑制剂治疗显著提高了高迁移率族蛋白 B1-1 (HMGB1) 59和 Ki-67 的表达水平(图4N-O),强调了 PCSK9 在调节肿瘤发生中的重要作用,这已通过该肺上皮类器官模型得到验证。
为了验证胆固醇强度与肺癌风险之间的相关性,对开滦队列(由中国唐山开滦地区的在职和退休人员组成,共计109884名男性参与者)的血液总胆固醇(TC)强度与肺癌发病率进行了分析。使用Cox比例风险回归模型评估了该队列中基线TC水平与肺癌风险之间的关联和非线性关系(图 4P)。校正年龄、教育程度、收入、吸烟和饮酒等因素后,与 TC 水平正常(160-180 mg/dl)的个体相比,较低 TC 水平(<160 mg/dl)和较高 TC 水平(>240 mg/dl)的个体肺癌风险的风险比 (HR)(95% 置信区间)分别为 1.34和 1.45(图 4P)。TC 水平较低或较高的个体均表现出较高的肺癌风险,这表明 TC 水平与肺癌风险升高之间可能存在关联,有待进一步的临床验证。
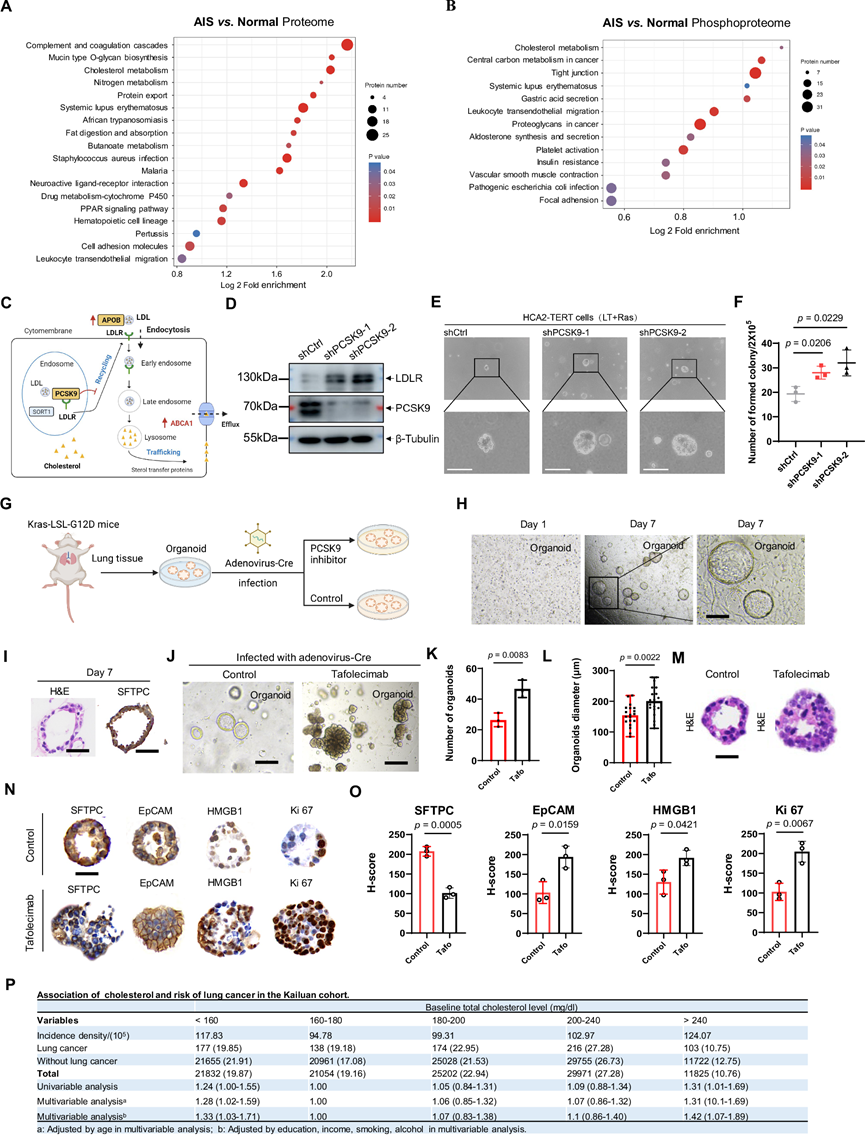
图4. 蛋白质组学表征揭示肺肿瘤前病变的发生。
(A-B) KEGG 富集分析。(C) 通过 pathview 分析胆固醇代谢通路和通路上靶分子的表达。(D) PCSK9 敲低对 HCA2-TERT 细胞中 LDLR 和 PCSK9 水平的影响。(E-F) 用编码 SV40 大肿瘤抗原 (LT) 和 HRAS V12 (Ras) 的载体与 shCtrl 或 shPCSK9 一起转染并在软琼脂中接种的 HCA2-TERT 细胞的贴壁非依赖性克隆形成检测。(G) 从 Kras-LSL-G12D 小鼠肺组织建立类器官的工作流程示意图。(H) 培养 7 天后类器官的明视野显微镜代表性图像。(I) 第 7 天的类器官的形态和组织学特征(表面活性蛋白 C:SPPTC)。(J) 用腺病毒-Cre 感染类器官。(K) 用腺病毒-Cre感染3 周后,所示组中形成的类器官数量。(L) 显示所示组中类器官直径量化的条形图。(M) PCSK9 抑制剂组和对照组中形成的类器官的形态。(N-O) 通过免疫组织化学 (IHC) 对 PCSK9抑制剂组和对照组中类器官的 SFTPC、EpCAM、HMGB1 和 Ki 67 的代表性图像和定量数据。(P) 2006-2014 年队列中109884 名参与者的总胆固醇与肺癌风险的关系。
05
内质网应激是 AIS 发展为 IAC 的标志,也是 LUAD 的药物靶点
KEGG 通路分析显示,与 NAT 相比,肿瘤中粘着斑的激活、内质网 (ER) 中的蛋白质加工、吞噬体、血小板活化和蛋白多糖在癌症通路中富集(图 5A)。为了进一步描绘从肿瘤前期到侵袭性 LUAD 的气道进展的进化轨迹,他们使用三个阶段之间的差异蛋白质和磷酸化位点以及糖肽进行了 mFuzz 聚类,并分别在蛋白质组学、磷酸化蛋白质组学和糖蛋白质组学水平上鉴定出四个不同的聚类。他们重点关注了聚类3中的蛋白质,其随着肿瘤进展呈现持续上升趋势,KEGG通路分析显示内质网蛋白质加工、真核生物核糖体生物合成、剪接体和溶酶体通路在聚类 3中富集。在磷酸化蛋白质组学水平上,聚类4中蛋白质的磷酸化修饰从AIS到MIA和IAC呈现逐步增加的趋势,且随着肿瘤进展呈现持续上升趋势。KEGG通路分析显示内质网蛋白质加工、凋亡和自噬途径在聚类4中富集。在糖蛋白质组学水平上,聚类2中蛋白质的糖基化修饰从AIS到MIA和IAC呈逐步增加的趋势,并随着肿瘤进展呈现持续上升的趋势。KEGG通路分析显示,内质网中的蛋白质加工、凋亡和自噬以及癌症通路中的蛋白多糖在聚类2中富集。
蛋白质二硫键异构酶 (PDI) 是一种调节二硫键的 ER 伴侣,在 ER 应激过程中诱导,是 ER 中蛋白质正确折叠所必需的。为了研究 PDI 抑制对抗癌作用的影响,他们招募了一种名为 CCF642 的小分子抑制剂来增加 ER 应激。首先,他们从 LUAD 建立了表现为 SSN 的患者来源肿瘤异种移植 (PDX) 模型,用于检测 CCF642 的体内功效。用 CCF642 治疗可减少小鼠的肿瘤生长并显著延长小鼠的生存期(图 5B-F)。此外,为了进一步研究 ER 应激过度激活在 PDX 模型(PDX-P2)中 PDI 抑制的抗肿瘤作用中的作用,他们使用 IHC 检测法评估了 ER 应激的可靠标志物,包括 PERK 和 ATF4。他们发现 CCF642 治疗的肿瘤中 PERK 和 ATF4 的表达明显高于对照组。他们之前采用患者来源的肿瘤样细胞聚类模型(PTC 模型)评估肺癌药物疗效,该模型在预测化疗效果方面表现出很高的准确性。他们建立了 17 名 LUAD 患者的 PTC 来评估 CCF642 的效果,观察到 CCF642 有效抑制了 PTC 中的肿瘤生长,并且这种效果具有浓度依赖性(图 5G)。此外,他们发现 CCF642 对异种移植 LLC 细胞的生长具有类似的抑制作用(图 5H-J)。
随后,他们用 1 μM CF642 处理培养的 LLC 细胞,观察到对细胞增殖的抑制作用(图 5K)。重要的是,当专门针对 PDI 家族成员 Pdia3(图 5L)时,始终观察到强烈的抑制作用(图 5M),而 Pdia3 的敲低对细胞凋亡没有明显影响。此外,在自噬抑制剂氯喹 (CQ) 的情况下,Pdia3 敲低均不会显著改变相对 LC3-II/LC3-I 蛋白水平,表明 Pdia3 对自噬没有明显影响。此外,与对照组相比,沉默 Pdia3 可显著抑制 LLC 异种移植瘤的生长速度并减轻 LLC 肿瘤的重量(图 5N-P)。总体而言,这些发现表明抑制 PDI 可以有效阻止肺癌的进展。
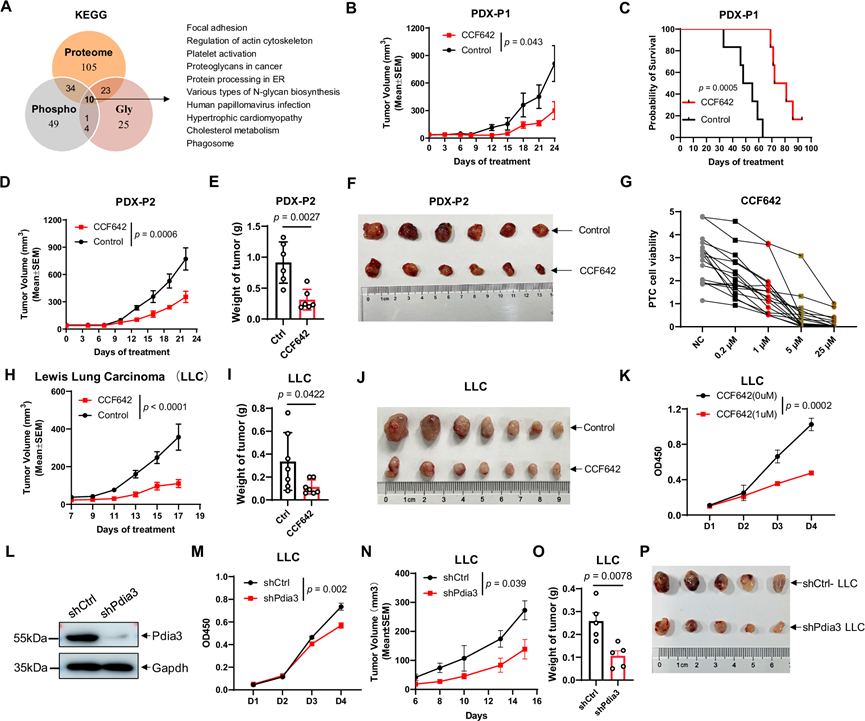
图5. 内质网应激是 IAC 进展的标志和药物靶点。
(A) 肿瘤与配对正常邻近组织 (NAT) 之间差异调节的蛋白质、磷酸化位点和完整糖肽以及通路。(B) 在 BALB / c 裸鼠中,未经处理或用 CCF642 处理的肺腺癌 (PDX-P1) 患者来源的异种移植瘤 (PDX) 形成的肿瘤的生长曲线。(C) CCF642 对中 PDX 小鼠生存的影响。(D) 在 BALB/c 裸鼠中,未经处理或用 CCF642 处理的另一种肺腺癌 (PDX-P2) 的PDX形成的肿瘤的生长曲线。(E) CCF642 对 PDX(PDX-P2)形成的肿瘤重量的影响。(F) BALB/ c裸鼠中未经处理或用 CCF642 处理的 PDX-P2 植入形成的肿瘤。(G) CCF642 作用下 17 个肺腺癌患者来源的肿瘤样细胞聚类 (PTC) 的药物反应概况。(H) C57BL/ 6小鼠中未经处理或用 CCF642 处理的同种异体移植WT的LLC 细胞形成的肿瘤的生长曲线。(I) CCF642对肿瘤重量的影响。(J-K) 在 C57BL/6 小鼠中,未经处理或用 CCF642 处理的同种异体移植 WT LLC 细胞形成的肿瘤CCF642 对 LLC 细胞增殖的影响。(L) Pdia3敲低对LLC 细胞中 Pdia3 水平的影响。(M) Pdia3 敲低对 LLC 细胞中细胞增殖的影响。(N) 在 C57BL/6 小鼠中,WT 和 Pdia3 敲低 LLC 细胞形成的肿瘤生长曲线。(O-P) Pdia3对形成的肿瘤重量的影响。
持续的ER应激是AIS进展为IAC的标志(图 5A)。为了进一步验证ER应激是否可以作为AIS进展为IAC的可靠生物标志物,他们从临床环境中收集了A1和A2肿瘤。生长曲线显示,通过评估CT扫描上的肿瘤大小,A1亚型肿瘤快速生长(图 6A),A2亚型肿瘤处于几乎稳定状态(图 6B-C)。随后,他们通过免疫组化 (IHC) 检测同时评估了可靠的内质网应激标志物(PERK、ATF4 和 eIF2S1 磷酸化)。他们发现 A1 亚型肿瘤中 PERK、ATF4 和 eIF2S1 磷酸化的表达显著高于 A2 亚型肿瘤,从而明确了内质网应激在肿瘤进展中的关键作用(图 6D-E)。此外,他们还在一个包含 110 例表现为 SSN 的 LUAD 患者的独立患者队列中验证了 PERK 的预测价值。在 110 例 LUAD 病例中,31 例(28.1%)表现出高 PERK 表达(定义为超过 10% 的 PERK 阳性细胞)。与 PERK 水平较低的患者相比,PERK 表达升高的患者术后复发率明显增加,表明 PERK 表达与侵袭性和进展趋势增强呈正相关。
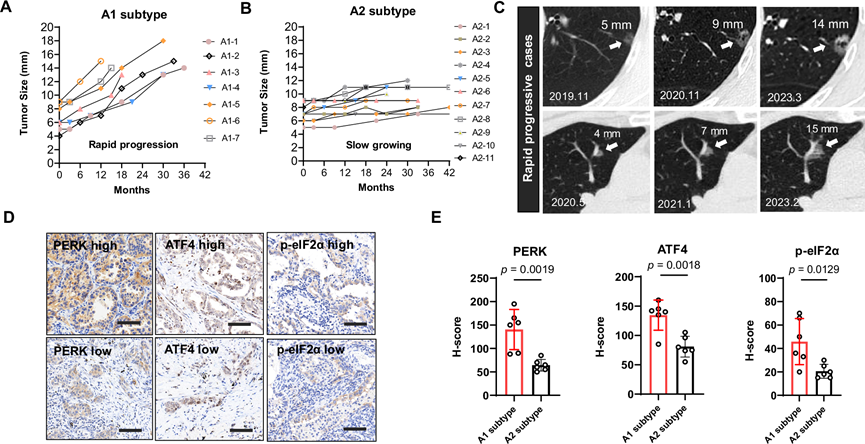
图6. 内质网应激是临床上 AIS 发展为 IAC 的标志。
(A-B) CT 扫描上 A1 和 A2 亚型肺腺癌的生长曲线。(C)代表性 CT 扫描。(D) A1和 A2 亚型肿瘤的 PERK、ATF4 和 p-eIF2S1 的代表性免疫组织化学 (IHC) 染色。(E) A1 和 A2 亚型肿瘤的 PERK、ATF4 和p-eIF2S1 的定量数据。
+ + + + + + + + + + +
结 论
本研究结合蛋白质组学、磷酸化蛋白质组学和糖蛋白质组学,分析了66例以SSN形式出现的LUAD,涵盖AIS、MIA和IAC 三个组织学阶段。值得注意的是,胆固醇代谢在肿瘤前AIS阶段受到异常调控。重要的是,靶向敲除PCSK9可促进LUAD的发生。此外,持续的内质网应激已证实是AIS进展为IAC的标志和可靠的生物标记。持续靶向促进内质网应激可显著延缓LUAD进展。本研究提供了SSN的全面蛋白质组学图谱,阐明了SSN的肿瘤发生和进展,并提出了LUAD的预防和治疗策略。
+ + + + +



