

 English
English文献解读|Sci Transl Med(17.1):用于精准肿瘤学的肝癌类器官的药物蛋白质组学表征
✦ +
+
论文ID
原名:Pharmaco-proteogenomic characterization of liver cancer organoids for precision oncology
译名:用于精准肿瘤学的肝癌类器官的药物蛋白质组学表征
期刊: Science Translational Medicine
影响因子:17.1
发表时间:2023.07.26
DOI号:10.1126/scitranslmed.adg3358
背 景
原发性肝癌是全球第三大癌症死亡原因,其中肝细胞癌(HCC)约占85%,其次是肝内胆管癌(ICC)和肝细胞胆管癌(CHC)。类器官模型有可能重现亲本肿瘤的生物学和药型特征。然而,仍然缺乏针对肝癌患者药物反应特征的综合药物-蛋白质组学分析和精准治疗的生物标志物研究。
实验设计
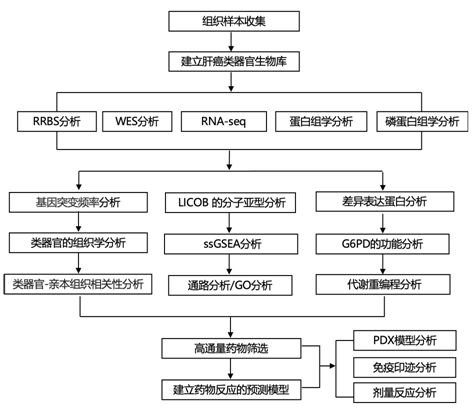
结 果
01

类器官概括了原发癌组织的组织学和分子特征
研究者团队收集了手术切除的原发性肝癌组织来生成肝癌类器官生物库(LICOB)(图1A),成功生成的类器官的 6 至 12 代用于进一步分析。6至12个成功生成的类器官用于进一步分析。对于每个类器官,用于多组学分析和药物筛选实验的培养间隔不超过1或2代。57例患者共建立65个类癌器官,包括HCC类器官 (HCCO)、ICC类器官 (ICCO)、CHC类器官 (CHCO),肝母细胞瘤类器官(HBO)。其中40例为乙型肝炎病毒(HBV)阳性,13例来自同一患者的2-4个肿瘤亚区(图1B)。这些类器官显示出癌症类型特异性形态(图1C),并概括了原始组织和癌症类型的组织学特征(图1D-F)。同样,HCCO、ICCO和CHCO/HBO分别表达HCC特异性、ICC特异性和两种类型的标记物(图1E-F)。
他们使用减少代表性亚硫酸盐测序(RRBS)、全外显子组测序(WES)、RNA测序(RNA-seq)和无标记蛋白质组学对这些类器官和亲本组织进行了蛋白质、基因组学分析,然后进行了高通量药物筛选。
首先,他们比较了类器官和配对癌症组织之间的体细胞突变、拷贝数变异(CNV)、DNA甲基化、转录组学和蛋白质组学特征。WES数据估计的类器官的肿瘤纯度接近100%,显著高于相应的癌组织。类器官及其亲本组织显示出相似的基因突变频率,包括HCC和ICC中反复突变的基因,包括TP53、KMT2C、RB1和PBRM1, HCC特异性突变的CTNNB1,以及ICC特异性突变的KRAS和BAP1(图1G)。同样,克隆分析显示的突变聚类和大多数基因的等位基因频率在类器官和原始组织之间高度一致。对于CNV、DNA甲基化、mRNA和蛋白质谱,配对的类器官组织样本的相关系数显著高于未配对的样本(图1H)。在无监督的转录组学分析中,类器官与来自癌症基因组图谱(TCGA)、临床蛋白质组学肿瘤分析联盟(CPTAC)的肝癌样本和配对组织形成聚类,而来自LIMORE的癌细胞系形成了不同的聚类,表明LICOB类器官比细胞系更好地代表肝癌。
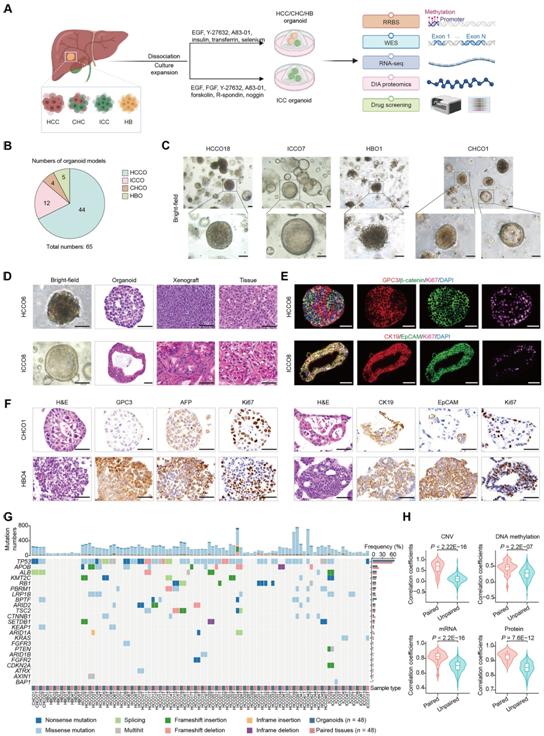
图1. LICOB的建立及与原发性肝癌的比较。
(A) 肝癌类器官药物蛋白质组学分析的流程示意图。(B) LICOB 中类器官模型的数量。(C) LICOB 的代表性明场图像。(D) 类器官、异种移植物及其亲本肿瘤的代表性苏木精和伊红 (H&E) 染色。(E-F) LICOB 中所示标记物的免疫荧光和免疫组织化学分析。(G) LICOB 和配对癌症组织的突变景观。(H) 不同组学中类器官与配对或未配对组织的 Spearman 相关系数。
02
LICOB 的多组学分类确定了四种亚型
他们根据五个组学数据集对类器官进行了共识聚类,并确定了具有不同分子特征的四种亚型(图2A)。所有的ICCO都归类为具有RAS信号和细胞连接升高的聚类3(图2A-B),这意味着聚类3具有ICC主导特征,因此表示为LICOB (L) -ICC。聚类2在细胞周期和有丝分裂原激活的蛋白激酶通路中富集,因此将其命名为L-PL,表示增殖亚型(图2A-B)。多能相关基因的DNA甲基化显著降低,肿瘤干细胞标志物CD44在L-PL中上调(图2A-B),L-PL扩增时间明显短于其他亚型。聚类1和聚类4分别为脂质代谢和药物代谢通路富集,因此分别标记为L-LM和L-DM(图2A-B)。L-LM表现出APOB突变积累,CPS1和PLA2G2A mRNA丰度升高,而L-DM表现出ALB和PLEC突变增多,UGT1A1和ALDH1A1蛋白丰度升高(图2A)。除ICCO外,在LICOB数据集中确定的亚型与之前基于HCC组织的聚类结果基本一致(图2C)。例如,L-PL与公开数据中的增殖亚型匹配良好,而L-DM更接近中间亚型(图2C)。
为了进一步探讨 LICOB 亚型的预后价值,他们利用 LICOB 聚类的蛋白质组学特征将 CPTAC 队列中的 159 名 HCC 患者分为三个亚组,发现 L-PL 的生存期最差,而 L-LM 显示最好的预后(图2D),表明 CPTAC 队列中增殖亚型和代谢亚型的患者分别具有最差和最好的生存率。药物代谢上调的L-DM表现出低于L-LM的中等生存率(图2D),这可能是由于两种亚型之间的代谢通量不同。与L-DM中的磷酸戊糖代谢和谷胱甘肽代谢相比,差异表达的基因证明L-LM中的糖酵解和脂质代谢通路上调(图2E-F)。这种独特的代谢程序可能会导致具有 L-DM 特征的患者预后较差。

图2. LICOB 的分子亚型。
(A) 基于揭示四种亚型的多组学数据的共识聚类。每列代表一个类器官样本,行表示分子特征。(B) 每个 LICOB 多组学聚类和每种类型组学数据中选定基因集的 ssGSEA 分数。(C) LICOB 中的 HCCO 亚型与之前患者 HCC 组织的亚型结果的比较。(D) CPTAC HCC 队列中总体生存的 Kaplan-Meier 曲线根据类器官亚型的多组学特征进行聚类。(E) 象限图描绘了与 L-DM 相比,L-LM 中转录组和蛋白质组同时检测到的 6729 个基因的改变。(F) L-LM 和 L-DM 之间GO分析。
03
G6PD 介导 L-DM 亚型的代谢重编程
为了探索潜在的治疗靶点,他们对之前报道的 HCC 中的可药物靶点进行了差异蛋白表达分析,发现与其他亚型相比,葡萄糖-6-磷酸脱氢酶 (G6PD) 在 L-DM 中显著上调(图3A-B)。G6PD mRNA 和蛋白质丰度在 LICOB 和 CPTAC HCC 数据集中高度相关。在CPTAC或TCGA HCC数据集中,高G6PD丰度患者的生存率显著低于低G6PD丰度患者,提示G6PD是一种促肿瘤因子(图3C)。
G6PD是与糖酵解平行的戊糖磷酸通路(PPP)中的限速酶,对于生成还原形式的烟酰胺腺嘌呤二核苷酸磷酸(NADPH)进行氧化还原调节和为核苷酸合成提供底物至关重要。单样本基因集富集分析(ssGSEA)表明,L-DM中药物/外源代谢、核苷酸代谢通路和生物氧化增强,而糖酵解基因下调并发生高甲基化(图3D-E)。因此,G6PD 的异常高表达可能会重新连接 L-DM 中从糖酵解到 PPP 的代谢。
与转录组学和蛋白质组学分析类似(图2F),代谢物富集分析显示,与L-DM中的谷胱甘肽和核苷酸糖代谢相比,L-LM中的糖酵解和糖异生增强。PPP通路的关键酶包括PGD、TKT和TALDO1在L-DM中表达上调,并与G6PD蛋白丰度呈显著正相关,而乙醇酸酶在L-DM中表达下调(图3F-G)。
G6PD敲除后,L-DM内HCCO8和HCCO12的增殖受到抑制(图3H-I)。在长期培养过程中,G6PD的敲低也阻碍了三维类器官的生长,表现为平均大小减小,小面积类器官比例增加(图3J)。正如预期的那样,G6PD敲除降低了NADPH和GSH的产生(图3K),表明增殖抑制可能是氧化还原稳态失调造成的。与L-LM类器官相比,L-DM类器官对选择性G6PD抑制剂(G6PDi-1)更敏感,表明靶向G6PD的治疗潜力(图3L)。MYC扩增也是L-DM的一个关键特征(图3M), MYC和G6PD的表达同时增强,两者之间呈正相关。MYC扩增分别与PPP通路和糖酵解通路蛋白表达呈正相关和负相关(图3N)。与NRF2通路在L-DM中的表达上调一致,G6PD转录激活因子NRF2在L-DM中的表达量在LICOB数据集中最高。因此,MYC扩增可能通过NRF2激活G6PD的转录,然后通过增强PPP通量来支持细胞生长和抗氧化防御,从而增强氧化应激下的生存和进展(图3O)。这些结果确定了G6PD的治疗价值,突出了多组学数据驱动治疗发现的可行性。
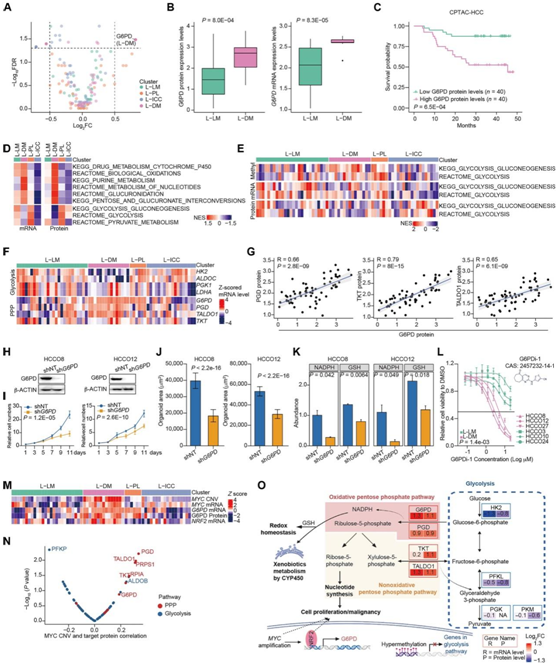
图3. G6PD 作为 L-DM 癌症类器官的潜在药物靶点。
(A) LICOB 亚型中潜在药物靶点的差异表达蛋白。(B) L-LM 与 L-DM 中的 G6PD 蛋白和 mRNA 表达。(C) 基于 CPTAC HCC 数据集中 G6PD 蛋白的下分位数和上分位数表达的 Kaplan-Meier 总生存曲线。(D) LICOB 聚类中指示路径的 ssGSEA 评分。(E) 糖酵解通路的 ssGSEA 评分。(F) 每个 LICOB 样品中 PPP 和糖酵解相关基因的 mRNA 表达。(G) G6PD 表达与每个所示蛋白质之间的 Pearson 相关性。(H) 使用G6PD沉默 (sh-G6PD ) 或对照 (sh-NT) 对来自 L-DM 的 HCCO8 和 HCCO12 中的 G6PD 进行免疫印迹分析。(I) 指定类器官的增殖。(J) 指定类器官的面积。(K) 箱线图展示了指定类器官中的指定代谢物。(L) L-LM 和 L-DM 类器官 G6PD 抑制的剂量反应曲线。(M) LICOB 亚型中选定的多组学特征。(N) MYC CNV 与糖酵解(蓝点)和 PPP 通路(红点)的蛋白质表达之间的 Spearman 相关性,并标记了显著相关的基因。(O) 示意图描绘了 G6PD 在 L-DM 代谢变化中的功能。
04
LICOB 药物反应的异质性与分子亚型相关
接下来,他们使用 LICOB 队列对 76 种美国食品和药物管理局批准的临床或实验开发药物和化合物进行了高通量药物筛选(图 4A)。通过计算曲线下面积 (AUC)、半最大抑制浓度 (IC50) 和最大效应 (Emax) 来确定类器官治疗反应。LICOB模型的治疗反应模式不同, HBV阳性的类器官通常比HBV阴性的类器官对化疗更耐药(图4B)。大多数类器官对针对相同靶点的药物表现出相似的反应,如DNA拓扑异构酶II(TOP2)抑制剂多柔比星和表柔比星,组蛋白去乙酰化酶(HDAC)抑制剂belinostat和vorinostat、溴结构域和超末端(BET) PROTAC抑制剂dBET6和ARV-771(图4C)。
几乎所有的类器官都显示出对olaparib的耐药性,而一些HBO和ICCO对talazoparib敏感(图4C),这表明有必要比较不同药物对相同靶点的疗效和潜在机制。在四种LICOB亚型中观察到不同的药物/化合物反应模式(图4D)。增生性亚型L-PL对靶向磷脂酰肌醇3-激酶(PI3K) -AKT-mTOR通路的抑制剂BEZ235和替西罗莫司(temsirolimus)敏感,而代谢性亚型L-LM和L-DM耐药。具体来说,L-PL对化疗药物的敏感性最高(图4D-E),在细胞周期通路上的评分高于其他亚型(图4F)。L-LM和L-DM对tivantinib的敏感性更高(图4D)。Tivantinib是一种选择性c-MET抑制剂,在2期试验中显示出对HCC有希望的效果。
他们在L-LM或L-DM中未观察到MET突变、扩增或mRNA或蛋白丰度升高。然而,MET激活所需的基因在两种亚型中均显示出较高的CNV以及较高的mRNA和蛋白丰度(图4G),包括MET配体HGF及其激活剂HPN、通过去泛素化稳定MET的USP8、MET激活所需的DOCK7,以及连接信号转导至细胞内通路的下游效应蛋白CRKL。因此,tivantinib的疗效可能与MET激活和功能基因的上调有关,而不是与MET表达有关。两项3期研究报告了tivantinib对c-MET高表达的HCC患者无效,这提示多组学生物标志物驱动的患者选择可能有助于识别tivantinib敏感亚群。几种抑制剂在L-LM和L-DM之间显示出不同的疗效(图4D)。L-DM对成纤维细胞生长因子受体(FGFR)抑制剂BGJ398和PD173074的敏感性最高,蛋白质基因组学分析显示该亚型通过相应基因的低甲基化、扩增、mRNA和蛋白丰度升高而激活FGFR通路(图4D-G)。
与HCC亚型相比,L-ICC类器官对酪氨酸激酶抑制剂耐药,这突显出HCC和ICC之间不同的药物应答模式。L-ICC内的药物反应也具有异质性,根据敏感性评分,ICCO2、ICCO5、ICCO10、ICCO11和ICCO12较其他ICCO普遍耐药,因此称为R-ICCO,其他称为S-ICCO(图4H)。他们根据R-ICCO和S-ICCO蛋白基因组特征将CPTAC队列中的ICC患者聚类,并确定了两个生存显著不同的亚组(图4I)。同样,通过在通路水平上比较L-ICC类器官与CPTAC队列中ICC组织的蛋白质组学特征,R-ICCO接近原始的最差生存亚型(S1)(图4J)。蛋白质基因组学分析表明,EGFR-酪氨酸激酶抑制剂(TKI)耐药特征包括TP53和KRAS突变、ERBB2和MAP3K3扩增、PTPN11和MAP3K2 mRNA丰度增强以及干扰素信号通路在R-ICCO中富集(图4K-L)。总之,这些结果表明LICOB中的异质性药物反应与特定的多组学特征相关。
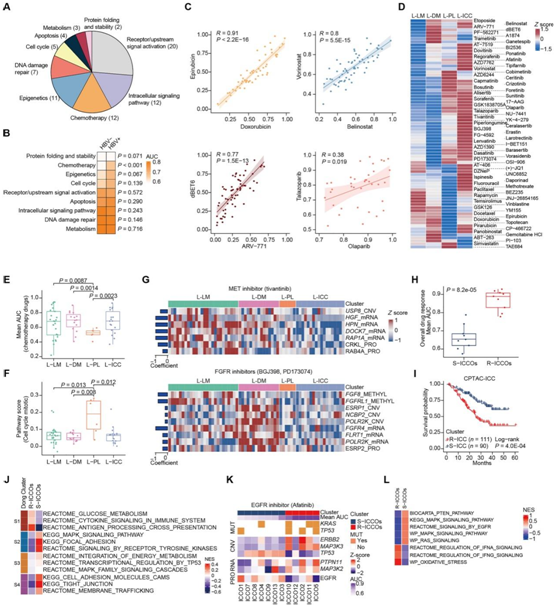
图4. LICOB 类器官中的异质药物反应。
(A) 76 种筛选药物的已知作用机制。(B) 每个药物类别的 HBV 阳性和 HBV 阴性患者的类器官之间的平均 AUC 值。(C) 具有共有分子靶标的药物的 AUC 值的代表性散点图。(D) 76 种药物的四种亚型的平均 AUC 值。AUC 值是按行归一化的Z分数。(E) 四种亚型化疗药物 AUC 值的比较。(F) 四种亚型中 Reactome 细胞周期有丝分裂通路的 ssGSEA 评分。(G) 与 FGFR 抑制剂(BGJ398 和 PD173074)或 MET 抑制剂 (tivantinib) 反应相关的多组学特征。条形图显示了药物 AUC 与每个聚类中特征值之间的 Spearman 相关性。对于BGJ398和PD173074,对相关系数进行平均。(H) S-ICCO 和 R-ICCO 之间平均 AUC 值的分布。(I) 基于 L-ICC 聚类特征的 CPTAC 队列中总体生存的 Kaplan-Meier 曲线。(J) 使用 CPTAC 队列中报告的代表性路径比较 L-ICC 中的 R-ICCO 和 S-ICCO 聚类。(K) 与阿法替尼 (afatinib) 反应相关的多组学特征。(L) 基于蛋白质组数据的 S-ICCO 或 R-ICCO 中指定基因集的 ssGSEA 分数。
05
利用蛋白质组数据建立药物反应的预测模型
为了识别有助于药物反应的稳健分子特征,他们使用多组学数据构建了弹性网络回归模型(图5A-B)。
在已批准的肝癌治疗药物中,预测的 AUC 与实测的 AUC 具有较高的相关性,其中二线药物瑞戈非尼(regorafenib)的预测结果最好,其次是一线药物乐伐替尼(lenvatinib)和索拉非尼(sorafenib)(图 5C)。对于索拉非尼,与较高类器官敏感性相关的特征包括MYEOV的高甲基化;EFNA2、TINAG和UPK3A的 mRNA 丰度较高;并降低IGF1R蛋白丰度(图5D)。MYEOV已证明可促进癌症进展和转移,但其在HCC中的功能尚不清楚。与索拉非尼耐药相关的多组学特征在酪氨酸和细胞内激酶通路中富集,这些通路包括IGF1/IGF1R、EGFR、成纤维细胞生长因子11 (FGF11)、RAS和细胞外信号调节激酶(ERK)信号通路(图5E)。此外,HDAC通路和内吞作用也与索拉非尼耐药相关。他们还应用弹性网络模型来预测癌症药物敏感性基因组学 (GDSC) 数据库中的索拉非尼反应,并发现预测的 AUC 与 HCC 细胞系中观察到的反应良好相关(图 5F)。进一步分析表明,乐伐替尼耐药与KRT19和NDN mRNA丰度升高以及血栓素通路富集正相关,与醇脱氢酶1C (ADH1C)、羧酸酯酶1 (CES1)、血管内皮生长因子(VEGF)通路相关蛋白丰度升高负相关(图5G-I)。虽然这三种药物都是多靶向酪氨酸激酶抑制剂,但与索拉非尼和瑞非尼不同,乐伐替尼主要通过靶向促血管生成分子VEGFR1-3和FGFR来抑制血管生成。
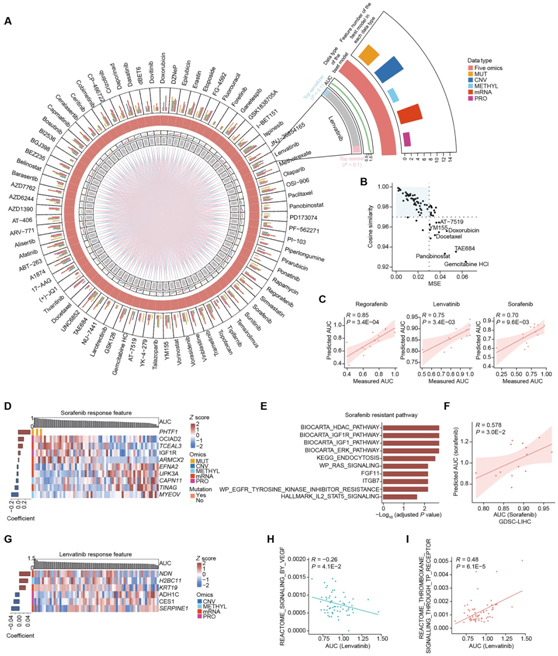
图5. LICOB 中的药物蛋白质组学分析。
(A) Circos 图显示 76 种药物中每种药物的弹性网络预测模型。第一个内圈代表每个LICOB样本的灵敏度;蓝色和粉色分别代表最敏感和最耐药的样本。第二个内圆表示每个类器官模型中特定药物反应的 AUC 值。第三个内圈表示显示出最佳预测精度的多组学或单组学数据类型,最外圈表示每个预测模型中的特征数量。(B)通过余弦相似度和MSE评估的药物预测精度的比较。(C) 指定药物的 LICOB 测试集中预测 AUC 值与测量 AUC 之间的 Spearman 相关性。(D) 索拉非尼具有最佳预测精度的多组学特征。左侧的条形图显示了模型中特征的系数。(E)代表索拉非尼耐药的多组学特征富集的通路。(F) GDSC 中乐伐替尼的预测 AUC 值与测量 AUC 之间的 Spearman 相关性。(G) 乐伐替尼具有最佳预测准确性的多组学特征。(H-I) LICOB 模型所示通路活性与 AUC 值之间的 Pearson 相关性。
06
确定促进乐伐替尼反应的联合方案
他们开发了一种基于网络的方法,使用蛋白质组数据预测 LICOB 队列中的候选药物组合(图 6A)。在76种测试药物的所有可能的药物组合中,他们预测乐伐替尼与YM155或替西罗莫司可能具有更好的组合效果(图6B)。替西罗莫司和乐伐替尼通过EGFR抑制剂耐药和转化生长因子-β信号通路相互作用,这些通路分别与乐伐替尼和替西罗莫司的敏感性呈负相关和正相关(图6B)。同样,替西罗莫司也列为tivantinib的首选补充药物。这些互补抑制也由(epi)基因组、CNV和转录组数据所证实(图6C)。与此一致的是,最近的一项高通量药物联合试验也显示了靶向受体酪氨酸激酶和PI3K-mTOR通路在乳腺癌、结肠癌和胰腺癌细胞系中的高度协同作用。
对乐伐替尼或替西罗莫司耐药的 HCC 类器官预计也对其组合敏感;即使是对乐伐替尼几乎没有反应的 ICCO,对于这种组合也往往有更高的分数(图 6D)。
为了验证这一预测,他们通过治疗四种预计对联合治疗有反应的选定类器官进行了比较研究。治疗后 6 小时,他们收集了类器官用于全磷酸化蛋白质组学分析,其中包括 23754 个量化的磷酸位点。乐伐替尼或替西罗莫司处理的类器官显示出两种药物所作用的通路(包括mTOR、VEGF和血小板源性生长因子通路)中涉及的磷酸化位点的独立下调,而与二甲基亚砜相比,乐伐替尼和替西罗莫司联合处理导致几乎所有上述磷酸化位点的下调(图6E),表明药物组合的有效性。其次,通过体外ICCO10、ICCO2、HCCO3、HCCO31实验验证,乐伐替尼单用对细胞增殖抑制作用有限,而与替西罗莫司联用则具有较强的协同抑制作用(图6F)。乐伐替尼单独使用可轻微降低MEK/ERK的激活,同时增强AKT的磷酸化,而与替西罗莫司联用可增强MEK/ERK的抑制,逆转AKT的激活(图6G),提示可能存在协同作用机制。与单独使用乐伐替尼和替西罗莫司治疗相比,具有HCCO31异种移植物的小鼠显示出明显的肿瘤生长抑制(图6H)。他们在体外类器官模型中验证了乐伐替尼和ganetespib预测的协同作用。值得注意的是,乐伐替尼+替西罗莫司在来自乐伐替尼耐药HCC患者的患者源异种移植(PDX)模型中显示出显著的肿瘤缩小效果(图6I),表明两种药物在体内的潜在协同作用。

图6. LICOB 中的药物组合预测和验证。
(A)药物组合预测的工作流程。(B) 乐伐替尼的药物通路网络图。橙色节点代表预测药物与乐伐替尼的组合效率,节点大小表示与乐伐替尼的组合分数。绿色节点表示连接每对药物组合的通路。(C) 乐伐替尼与其他药物联合使用的预测评分。点的大小代表不同组学数据贡献的分数。(D) 所有 LICOB 样本的乐伐替尼和替西罗莫司组合的预测评分。(E) 热图显示用指定药物处理的类器官中磷酸化位点的标准化丰度。(F) 热图显示 HCCO31 中乐伐替尼-替西罗莫司组合的剂量反应矩阵。(G) MEK-ERK 级联和 AKT 激活的免疫印迹分析。(H-I) HCCO31 和乐伐替尼耐药 PDX 模型异种移植模型中药物对肿瘤生长的功效。
+ + + + + + + + + + +
结 论
本项研究建立了一个源自患者的LICOB,通过多组学分析(包括基因组、表观基因组、转录组和蛋白质组分析)确定各种肝癌类型的组织学和分子特征。LICOB 的蛋白质基因组分析确定了与患者预后相关的增殖和代谢类器官亚型。高通量药物筛选揭示了与特定多组学特征相关的每种亚型的独特反应模式。通过对 LICOB 药物蛋白质组学数据的综合分析,本项研究确定了与药物反应相关的分子特征,并预测了个性化患者治疗的潜在药物组合。mTOR 抑制剂替西罗莫司和多靶点酪氨酸激酶抑制剂乐伐替尼的协同抑制作用在类器官和患者来源的异种移植模型中得到了验证。本项研究为肝癌生物学和药理学依赖性的研究提供了丰富的资源,并可能有助于实现功能性精准医学。
+ + + + +



