

 English
English文献解读|Cancer Cell(50.3):胶质母细胞瘤进化的综合蛋白质组学特征
✦ +
+
论文ID
原名:Integrated proteogenomic characterization of glioblastoma evolution
译名:胶质母细胞瘤进化的综合蛋白质组学特征
期刊:Cancer Cell
影响因子:50.3
发表时间:2024.01.01
DOI号:10.1016/j.ccell.2023.12.015
背 景
胶质母细胞瘤(GBM)是成人中最常见、最致命的原发性脑肿瘤,尽管人们不断努力创新治疗方法,但中位生存期仍不足 15 个月。由于肿瘤异质性的复杂性以及对治疗后进化动态的了解有限,复发性 GBM 在治疗上仍未得到解决。
实验设计

结 果
01

纵向胶质母细胞瘤的分子轨迹
为了研究驱动GBM进化复杂分子轨迹的潜在机制,研究者团队收集了来自5个不同机构的123例匹配的GBM纵向标本。其中,122个通过全外显子组测序(WES)(其中91个有匹配的血液来源的正常DNA),86个通过全转录组测序(WTS),105个通过基于质谱的蛋白质组学来进行蛋白质和磷酸化蛋白定量(图1A)。
他们观察到异柠檬酸脱氢酶 1 (IDH1) 突变体和IDH1野生型 GBM复发后肿瘤突变负荷显著增加,而非整倍体分数没有表现出显著差异(图1B)。接下来,他们研究了GBM进化过程中主要驱动突变和拷贝数改变的发生情况。首先关注了构成胶质母细胞瘤核心致癌通路的驱动基因,包括受体酪氨酸激酶(RTK)-RAS信号通路、磷脂酰肌醇-3-激酶(PI3K)、p53、染色质修饰和细胞周期通路,IDH1突变在时间进化过程中表现出一致的稳定性(图1C)。同样,在未治疗和复发的肿瘤中,EGFR的染色体扩增、CDKN2A缺失以及PTEN、ATRX和TP53的体细胞突变高度保守。然而,他们发现了在复发性肿瘤中经常丢失的几种遗传改变,包括CDK6、MDM4和CCND2的染色体扩增(图1C-D)。
由于这些驱动因素主要涉及细胞周期和增殖动力学,他们假设复发性胶质母细胞瘤不能通过增强细胞周期进展和增殖的传统机制来维持。为了评估拷贝数变异(CNV)对肿瘤进展不同阶段顺式基因表达[功能性拷贝数变异(fCNV)]的功能性影响,他们对mRNA表达和CNV进行了综合分析。从原发性肿瘤到复发性肿瘤,主要的 GBM 驱动基因改变(包括CDKN2A、TP53、EGFR和PTEN)得以维持。相反,原发性和复发性 GBM 中的几个基因发生了差异性改变。诊断时更频繁改变的基因包括CDK6和MET癌基因(扩增)以及NF1肿瘤抑制基因(突变/删除)(图 1 D)。尽管数量较少,但在复发性肿瘤中PI3KR1的体细胞突变或缺失的频率较高。将综合分析扩展到非驱动基因时,他们发现参与细胞周期、有丝分裂和 DNA 修复活动的基因(ERCC2、HUS1、PDGFA、RCF2和 BRAT1)的功能扩增在 GBM 中显著富集。
相比之下,复发性GBM的标志是神经元分化促进基因(包括SGK2、LIN7B、STX1A和CDK5)的功能性扩增(图1E),这表明细胞增殖可能是治疗前GBM肿瘤扩张的驱动力。然而,经过标准治疗后,复发性GBM获得了不同于细胞周期和增殖的能力,可能涉及神经元活动的选择。
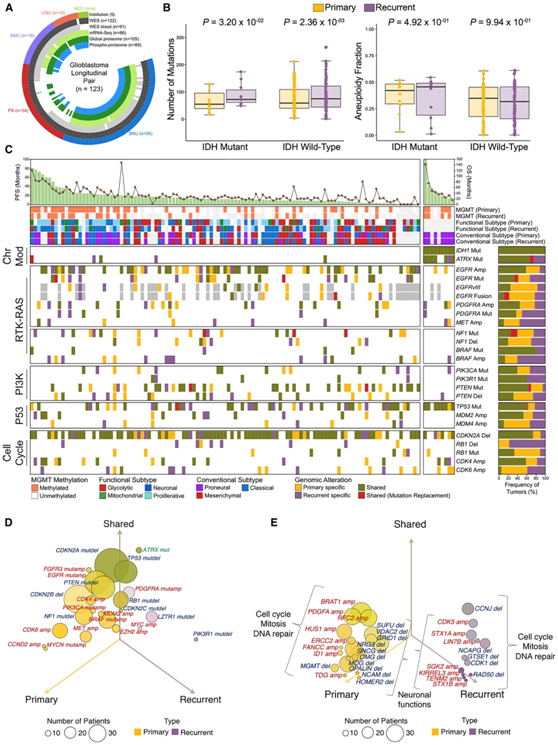
图1. 纵向胶质母细胞瘤的分子轨迹。
(A) 分析平台总结以及每个分子平台分析的匹配纵向 GBM 患者样本数量。(B) IDH 突变体和 IDH 野生型原发性和匹配的复发性 GBM 之间的肿瘤突变负荷和非整倍性分数的比较。(C) 按致癌途径分组的 GBM 驱动基因的体细胞基因组景观。(D) 气泡图显示f CNV 和 GBM 驱动基因非同义突变的频率,在原发性和复发性(紫色,右)和共有性(绿色,上)中完全原发(黄色,左)。(E) 气泡图显示了仅原发性(黄色,左)、仅复发性(紫色,右)以及原发性和复发性 GBM 之间共有的所有基因的fCNV 和非同义突变的频率(绿色,上)GBM 肿瘤。
02
EGFR基因组改变对复发性 GBM 下游信号传导的影响
先前的研究已经确定了 GBM 核心致癌途径中反复出现的基因组畸变,这些畸变构成了复杂的典型信号转导。其中,经常发现编码 RTK 的基因中的染色体扩增。当他们在 GBM 进化过程中在蛋白质水平上研究这些关键的致癌信号通路时,发现 FGFR2/3、NTRK2/3 和B-raf (BRAF)蛋白主要富集于复发性肿瘤中,而 EGFR、原发性肿瘤中 CDK4 和 CDK6 蛋白增加(图 2A)。这些致癌蛋白的积累与基因拷贝数和/或 mRNA 水平的相应变化无关,这表明 GBM 进化过程中这些蛋白激酶的转录后调节机制。EGFR的基因组改变是 GBM 肿瘤发生的主要因素之一。他们确定了三组不同的患者:一组在原发性肿瘤和复发性肿瘤中均表现出EGFR改变,而在其他两组中,复发性肿瘤中EGFR改变要么丢失,要么增加(图 2B)。正如预期的那样,复发时EGFR改变的丧失与 EGFR 蛋白的减少相关(图 2C)。在复发时维持EGFR改变的肿瘤中也检测到 EGFR 丰度和磷酸化的减少,这表明翻译后机制在进化过程中发挥作用以减弱 EGFR 信号传导(图 2D)。
EGFR 通过两个主要下游信号传导(PI3K-AKT-PTEN-mTOR 和 RAS-RAF-MEK-ERK 途径)发挥致癌效应。他们发现 EGFR 和GAB1与AKT1和AKT2蛋白之间存在统计学上显著的正相关性,但与 BRAF 和 MAPK1/3 蛋白之间存在负相关性(图2E)。他们发现磷酸化 EGFR(Y1197、S1166)在维持EGFR基因改变的复发性肿瘤中以及在那些在复发时病灶消失的患者(图2D)。磷酸化GAB1(S402、T503)在复发时显著减少(图2F)。这些结果表明,EGFR 信号传导在复发性 GBM 中很大程度上失活,这可能是进化过程中 EGFR 蛋白减少的结果。
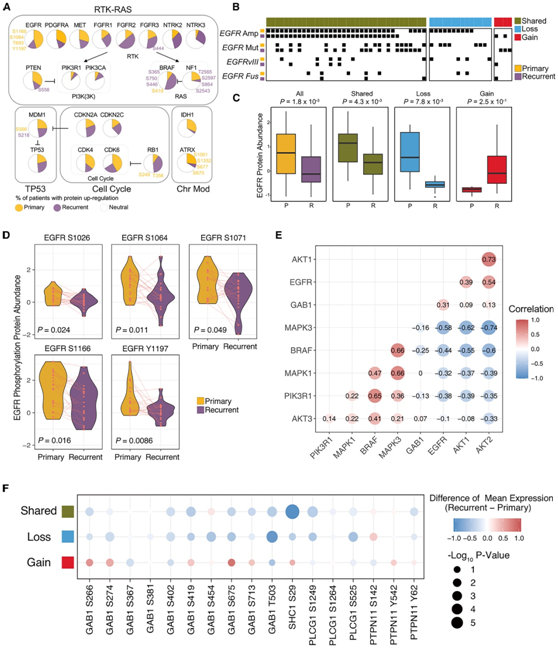
图2. EGFR 基因组改变对复发时下游信号传导的影响。
(A) 关键致癌 GBM 通路中上调蛋白的分析。(B) EGFR改变的体细胞基因组景观。(C) 相应患者组中匹配的原发性 (P) 和复发性 (R) 肿瘤之间 EGFR 蛋白丰度的比较。(D) “共有”组中原发性和复发性 GBM 患者之间 EGFR 位点磷酸化丰度的小提琴图。(E) EGFR 蛋白丰度与 PI3K-AKT-PTEN-mTOR 和 RAS-RAF-MEK-ERK 下游途径关键调节因子的相关图。(F) 点图显示比较复发性和原发性 GBM 时 EGFR 底物中磷酸化丰度的变化。
03
纵向 GBM 的蛋白质组学特征揭示了复发时神经元活性的增加
为了识别复发性 GBM 中活跃的生物学特征,他们分析了匹配 GBM 对的差异蛋白质组和磷酸蛋白质组,发现了 377 种蛋白质和 1820 种磷酸化蛋白质在复发性 GBM 中增加,包括SNAP25、TUBB4A、NELF、DAAM1/2、STMN1 和 MBP(图 3 A)。通路富集分析显示,复发性肿瘤的特征是参与神经元结构和功能以及突触形成(突触前和突触后活动)的蛋白质和磷蛋白的升高。在复发性 GBM 中显著过表达的生物通路包括“离子通道活性”、“动作电位”、“突触可塑性”、“神经递质运输”、“神经元投射”、“神经元分化”和“轴突”。这些途径富含了关键的结构神经元蛋白或参与神经元活动的蛋白质,即电信号和化学信号的传递。诊断时的 GBM 主要富含 DNA 复制激活、细胞增殖和丝裂原激活蛋白激酶 (MAPK) 调节以及细胞外基质功能和 EGFR 信号传导(图 3B-C)。
接下来,为了研究蛋白质调控与 GBM 进展的转录控制的相关性,他们整合了转录组、蛋白质组和磷酸化蛋白质组数据,以鉴定复发性肿瘤与原发性肿瘤中差异丰度的蛋白质和磷蛋白,显示 mRNA 的变化。接下来,为了研究蛋白调控与转录控制在GBM进展中的相关性,他们整合了转录组、蛋白质组和磷酸化蛋白质组数据,以鉴定在复发和原发肿瘤中表现出mRNA一致/不一致变化的蛋白和磷蛋白。虽然复发性GBM中大多数蛋白的相应mRNA有一致的变化(PLP1、GFAP、FBXO2、STMN4),但许多分子显示了mRNA-蛋白解离(SYT4/11、DAAM1/2、BRAF、EGFR及其底物GAB1)(图3D)。生物通路富集分析显示,复发时增加的蛋白和磷酸化蛋白与突触形成、GTP酶活性和氧化磷酸化显著相关。相反,与细胞分裂、mRNA加工和转录相关的蛋白质和磷蛋白在原发性GBM中更为丰富(图3E)。
然后,他们探索了遗传改变对复发时整体和磷蛋白丰度的功能性影响,包括顺式作用(同源基因产物)和反式作用(其他基因产物)。他们发现顺式对EGFR有很强的影响,在多个位点(S1071, S1166和T693)的总蛋白和磷酸化丰度在复发时显著降低(图3F-G)。这些结果表明,GBM中的EGFR改变有助于肿瘤的起始,但不再是肿瘤进化所必需的。在反式作用水平上,具有多种GBM驱动基因改变(如EGFR、PDGFRA、PTEN和TP53)的复发性肿瘤表现出RAS通路成员(NTRK2/3、BRAF和MAP2K1/2)、WNT通路成员(DAAM1/2)和参与突触形成的蛋白(PRKCZ、SNAP25、SYN1和GRIA2)的蛋白和磷酸化蛋白水平升高(图3F-G)。这些研究结果表明,复发性胶质母细胞瘤具有独特的神经元信号程序特征,这可能是由RAS和WNT信号通路的激活所驱动,并有助于抑制细胞增殖机制,从而导致原发性胶质母细胞瘤的扩增。
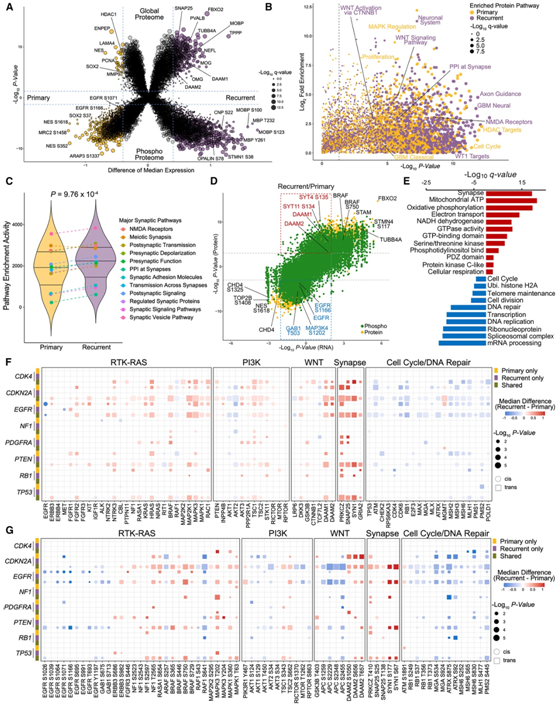
图3. 纵向 GBM 的蛋白质组学特征揭示复发性 GBM 中神经元活动的增加。
(A) 具有不同丰度的蛋白质(上)和磷蛋白(下)的火山图。(B)使用 MSigDB 中可用的所有基因集对原发性和复发性 GBM 之间的蛋白质和磷蛋白进行单样本基因集富集分析 (ssGSEA)分析。(C) 匹配的原发性肿瘤和复发性肿瘤之间主要突触通路活性的比较。(D) 散点图显示复发性 GBM 和原发性 GBM 之间 mRNA、蛋白质和磷蛋白表达/丰度的比率显著变化。(E) 对 (D) 中红色和蓝色象限的蛋白质和磷蛋白进行 GO 分析。 (F-G)主要基因组改变的顺式和反式影响,包括突变和 CNV 对蛋白质和磷蛋白水平的影响。
04
综合多组学亚型分析显示复发时神经元型GBM亚型富集
他们使用来基于通路的GBM分类法,研究了基因组学、转录组学、蛋白质组学和磷酸化蛋白质组学,以及聚类匹配的原发性和复发性GBM配对。该分类法将四种肿瘤亚型分为两个功能分支,神经发育分支[增殖/祖细胞(PPR)和神经元(NEU)]和代谢分支[糖酵解/多元代谢(GPM)和线粒体(MTC)]。他们发现,每个平台都富集了定义每个功能亚群的分子元件,并重现了每个GBM亚型的主要生物活性(图4A)。
然后,他们量化了匹配的原发和复发肿瘤的亚型转换频率,发现复发性GBM表现出显著较低的PPR频率和较高的NEU亚型频率(图4B)。为了确定基因改变的潜在影响,他们比较了原发性非NEU和复发性NEU GBM的驱动和非驱动基因改变。主要的GBM驱动因子如CDKN2A/B、EGFR、TP53和PTEN在原发和复发肿瘤中的发生率相似。与神经元分化和突触活动相关的基因如BRAF、STX1A、KCNN1、SCN1B和ACTL6B出现fCNV扩增,与细胞周期、有丝分裂和DNA修复相关的基因如CHEK2、ASPM、CENPW、CDK1和MDC1出现fCNV缺失。相反,原发肿瘤主要表现为参与细胞周期、有丝分裂和DNA修复功能的基因的fCNV优先增加,以及控制神经元分化和特化神经元功能的基因的fCNV缺失(图4C)。
GBM 中的化疗耐药很大程度上归因于MGMT 启动子甲基化抑制去甲基化酶O6-甲基鸟嘌呤-DNA 甲基转移酶 (MGMT) 的表达。当他们探究 MGMT 甲基化或 MGMT 未甲基化的原发性 GBM 在复发时是否表现出向特定功能亚型的优先转变时,48.6% 的 MGMT 未甲基化的原发性 GBM 复发为神经元肿瘤。一致地,复发时的 C2 亚型肿瘤表现出 MGMT 蛋白增加和 MMR 蛋白下调,包括MSH2、ML1 和 PMS2(图 4D)。相反,以 C1 或 C3 亚型复发的肿瘤没有表现出显著差异。蛋白质失活(可能通过蛋白质降解)也可能是导致 GBM 复发的重要机制(图4E)。
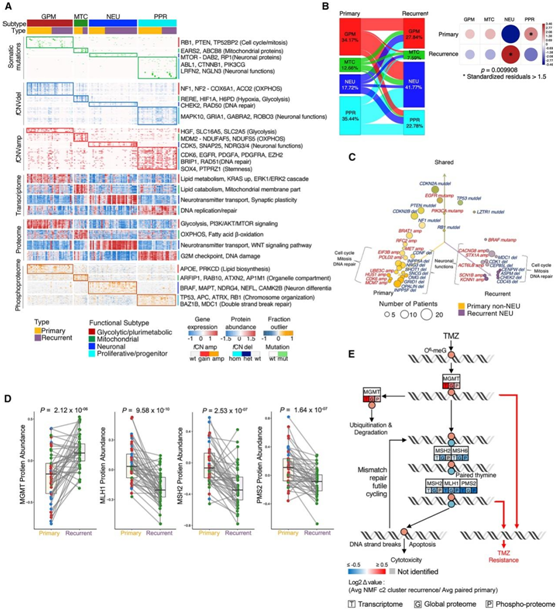
图4. 综合多组学聚类揭示了诊断时 GBM 向复发时神经元状态的转变。
(A) 根据原发性和复发性 GBM 基于蛋白质组的功能分类,与每个亚型显著相关的多组学特征的热图。 (B)原发性和复发性 GBM 的基于蛋白质组的功能亚型。(C) 与原发性或复发性肿瘤显著相关的基因中含有f CNV的原发性非神经元与复发性神经元匹配的 GBM 对的气泡图频率。(D)匹配的原发性肿瘤和转变为 C2 型的复发性肿瘤之间MGMT 、MSH2 、MLH1和PMS2蛋白丰度的比较。(E)错配修复(MMR) 编码蛋白下调导致替莫唑胺(TMZ)耐药的图示。
05
单细胞分析和功能实验模型揭示复发性肿瘤细胞中神经元活性的丰富
为了评估复发时神经元和突触活动的丰富是否是肿瘤细胞固有的而不是源自微环境,他们对四对原发性和复发性肿瘤组织样本进行了单细胞转录组分析(scRNA-seq)。他们首先根据 inferCNV 推断出的每个细胞中的大规模染色体 CNV 区分了 4228 个恶性细胞和 12258 个非恶性细胞(图 5A-B)。UMAP揭示了八种主要的非恶性细胞类型,它们具有高表达的不同标记基因,包括少突胶质细胞、骨髓细胞、T 细胞、星形胶质细胞、少突胶质细胞祖细胞 (OPC)、内皮细胞、周细胞、和神经元(图5C)。对比分析原发和复发GBM的肿瘤微环境(TME)发现,在复发肿瘤中,少突胶质细胞和神经元增多,髓样细胞和T细胞减少(图5D-E)。
在肿瘤细胞状态方面,他们基于通路功能状态分析了复发性GBM的进化轨迹,发现GBM进化的标志是PPR和MTC的降低和NEU状态的增加(图5F)。相反,其他胶质瘤细胞状态分类器,包括NPC样、OPC样、AC样和MES样状态在原发和复发GBM之间没有发现显著变化(图5F)。PPR和NEU细胞状态活性的计算证实,以牺牲干/祖细胞样活性为代价,神经元功能整体增加(图5G)。最后,他们在对应四种转录组学状态的四象限图中,计算了每个单细胞的“简单性评分”,作为状态强度的连续测量。肿瘤的象限/状态聚类表明神经元活动是复发性肿瘤的主要生物学特征(图5H)。

图5. 单细胞分析揭示复发性 GBM 的TME中肿瘤固有神经元状态以及神经元和少突胶质细胞的增加。
(A) 来自匹配的原发性和复发性肿瘤(4 名患者,8 个肿瘤)的单细胞的 UMAP 图,根据恶性或非恶性细胞亚群(左)和原发性或复发性状态(右)着色。(B) 根据 inferCNV 预测为胶质母细胞瘤标志的 7 号染色体(左)和10 号染色体(右)中基因拷贝数中位数着色的单细胞 UMAP 图。 (C) 根据恶性和非恶性细胞类型着色的单细胞 UMAP 图(左)和相应标记基因的平均基因表达(右)。(D) 条形图显示分析的每个肿瘤中的细胞类型组成。(E) 原发性和复发性 GBM 之间非恶性细胞类型的富集。(F) 基于功能途径(上)和神经胶质瘤细胞状态(下)的肿瘤细胞类型分布。(G) 箱线图显示来自原发性和复发性 GBM 的每个单独肿瘤细胞的神经元和增殖祖细胞状态富集评分 (NES)。(H) 功能性肿瘤细胞状态富集分数的二维图。
为了通过实验验证关于复发性 GBM 的基本组成部分(即神经发生和突触形成)的发现,他们使用共培养系统开发了体外模型。在该系统中,他们将带有 GFP 标记的匹配的原代和复发性 GBM 患者来源的肿瘤细胞 (PDC) 与小鼠皮质神经元共培养。结果表明,通过对 synapsin-1(突触前标记物)进行定量免疫荧光和免疫印迹分析进行评估时,与原发性肿瘤相比,复发性 GBM 细胞的特点是前突触数量增加(图 6 A)。与原发性肿瘤细胞相比,复发性肿瘤细胞每个细胞产生大量突触,并表现出更长的神经突(图6B)。此外,他们使用SOX2(GBM 肿瘤标记物)和 DAMM1(神经元发育)标记物对匹配的原发性和复发性 GBM 样本进行了双重免疫荧光分析,从原发性肿瘤的核心和浸润性周围选择了区域。有趣的是,无论分析哪个区域,在原发肿瘤的两个区域中几乎检测不到 DAMM1 的表达。相反,复发肿瘤在肿瘤核心表现出较高水平的DAMM1表达,在浸润边缘观察到显著增加(图6C-D)。这些结果证明了神经发生和突触形成作为复发性 GBM 的关键标志。
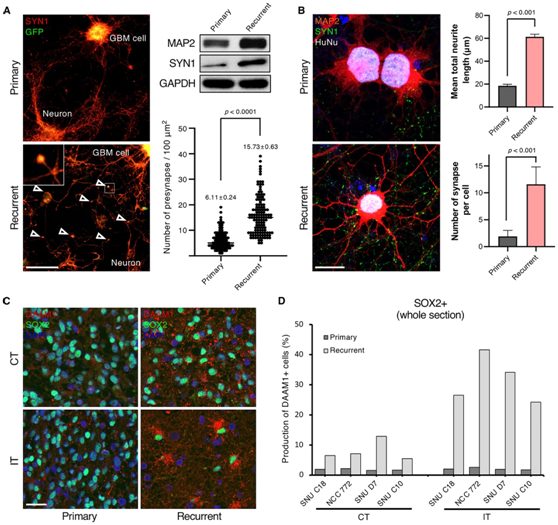
图6. 复发性 GBM 突触活性增强的功能验证。
(A) 配对的患者来源的 GBM 细胞(原发性和复发性)和小鼠皮质神经元细胞的共培养。(B)免疫荧光染色。(C-D) 患者 GBM 组织标本中 DAAM1(红色)和 SOX2(绿色)抗体免疫染色的代表性图像和定量。
06
使用体内模型系统对复发时的神经元转变进行建模,并以 BRAF 作为治疗靶标进行临床前测试
为了研究神经元复发 GBM 进化轨迹中转录组、蛋白质组和磷酸蛋白质组的动态调节,他们使用治疗后经历神经元转变的患者的肿瘤细胞生成了病人来源的异种移植瘤(PDX)模型(图7A),替莫唑胺(TMZ)处理显著延长了小鼠的总体存活率(图7B)。为了表征每个时间点,他们探究了转录组学、蛋白质组学和磷酸蛋白质组学数据,以识别独特的多组学特征。初始时间点(T7、T18 和 T27)表现出干性活性、细胞增殖和染色质重塑增加(图 7C)。在 T34(开始 TMZ 处理后 34 天),他们观察到最明显的神经元特征,特别是与突触结构和组织相关的特征。使用无监督共调节聚类的独立分析揭示了两个不同的蛋白质和磷酸位点聚类,它们在 T34 处显著分支(图 7D-E)。在T34降低的C1显著富集于细胞周期蛋白,氧化磷酸化,MYC和E2F靶点。相反,在T34富集的C2富集了参与神经元功能的蛋白和通路,如神经递质释放、多巴胺能突触和钙信号(图 7D-E)。免疫荧光分析证实,在TMZ处理的PDX中,与溶剂处理的肿瘤相比,T34的DAMM1表达增加(图 7 F)。
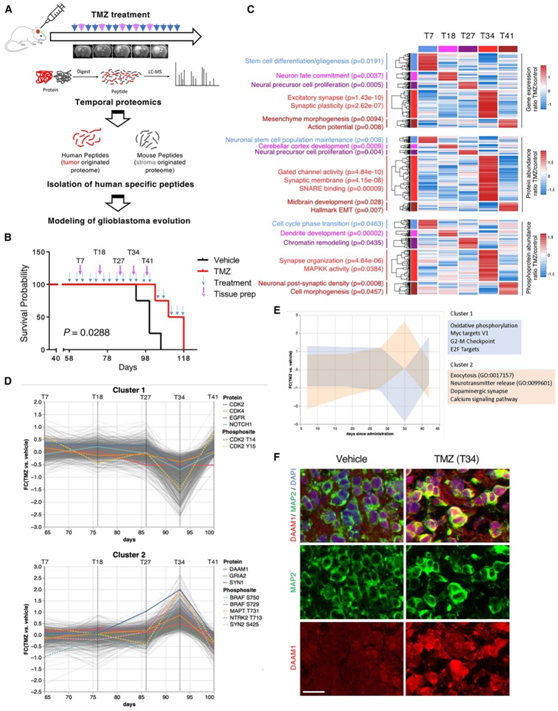
图7. 复发时神经元转变的体内模型。
(A) PDX 模型中 TMZ 治疗耐药性的蛋白质组学分析和建模实验设计概述。(B)生存分析。(C) 用 TMZ 多次处理的 PDX-GBM 的转录组(上)、蛋白质组(中)和磷酸化蛋白质组(下)的热图。(D) 对比 TMZ 和赋形剂治疗的 PDX 时间蛋白质组学数据的共调节聚类揭示了到给药后 35 天时具有相反表达模式的两个不同的蛋白质聚类。(E) (C) 中两个共表达蛋白质聚类的通路富集分析。(F) TMZ 和媒介物处理的 PDX 模型中 MAP2 和 DAAM1 的代表性多重免疫组织化学。
为了确定驱动复发性 GBM 神经元转变的潜在治疗靶点,他们使用 SPHINKS 重建了原发性复发性 GBM 特异性激酶-磷酸位点相互作用网络,SPHINKS 是一种最近开发的工具,集成了蛋白质组学和磷酸化蛋白质组学谱,以构建激酶-磷酸化相互作用组-根据所有样品之间相互作用的强度对底物对进行评分。
丝氨酸/苏氨酸蛋白激酶 BRAF 成为基于多组学的聚类 C2(主要由神经元复发性 GBM 组成)和基于功能分类器的 NEU 亚型中最活跃的激酶之一(图8A-B)。他们评估了两种 BRAF 抑制剂vemurafenib和dabrafenib对GBM 细胞神经突生长的治疗效果。用 1 μM 浓度的 BRAF 抑制剂处理会导致复发 GBM 细胞中的神经突缺陷(图 8 C)。此外,vemurafenib显著降低复发性 GBM 细胞中的 MAPK1/3 磷酸化,但对原发性肿瘤细胞没有影响(图 8D)。此外,vemurafenib与 TMZ 的组合显著延长了 GBM PDX 模型的总生存期并减小了肿瘤大小(图 8E-F)。这些发现表明,BRAF 在 GBM 进化过程中驱动神经元属性向神经元状态发展,并且 BRAF 抑制剂可以作为有用的治疗手段来预防与复发性 GBM 耐药性相关的神经元转化。
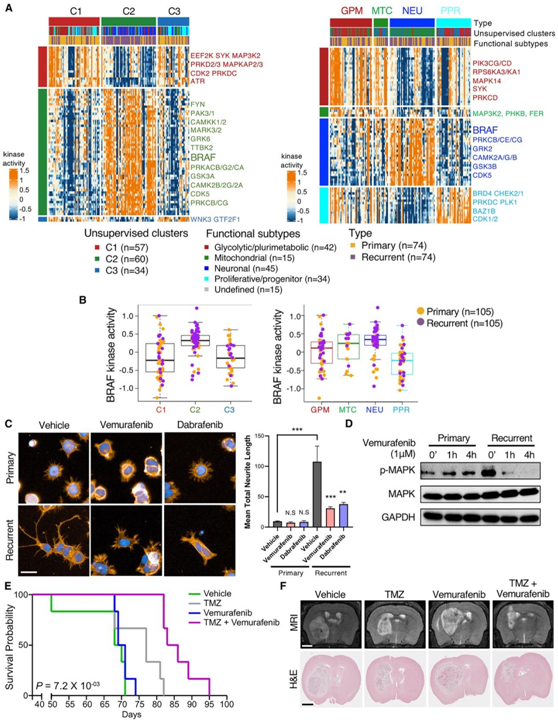
图8. 鉴定 BRAF 作为 GBM 进化的关键治疗靶点。
(A) 在每个无监督的基于多组学的(左)和功能性的(右)GBM 亚型中具有差异活性的主激酶热图。(B) 箱线图显示每个单独肿瘤中的 BRAF 活性,根据基于无监督多组学(左)或功能性 GBM 亚型(右)进行分层。(C) 代表性图像(左)和量化(右)在用载体、100 nM vemurafenib 或 100 nM dabrafenib (PMZ) 处理的成对 GBM 细胞中进行扩散测定。(D) 用维莫非尼处理的配对 GBM 细胞中 MAPK 信号相关蛋白的免疫印迹分析。(E)生存分析。(F) (E) 中小鼠大脑的代表性 MRI 和 H&E 切片。
+ + + + + + + + + + +
结 论
本项研究对 123 对纵向胶质母细胞瘤进行了综合蛋白质组学分析,并通过激活复发性肿瘤中的神经元转变和突触发生途径来识别诊断和替代时的高度增殖细胞状态。蛋白质组学和磷酸化蛋白质组学分析表明,复发时向神经元状态的分子转变以WNT/平面细胞极性(PCP)信号通路和BRAF蛋白激酶的翻译后激活为标志。PDX模型的多组学分析反映了相似的进化轨迹模式。B-raf 原癌基因 (BRAF) 激酶的抑制会损害复发性肿瘤细胞的神经元转换和迁移能力,这是治疗后进展的表型标志。替莫唑胺(TMZ) 与 BRAF 抑制剂vemurafenib的联合治疗可显著延长 PDX 模型的生存期。这项研究提供了对胶质母细胞瘤进化和治疗耐药性的生物学机制的全面见解,突出了有前景的临床干预治疗策略。
+ + + + +



