

 English
English文献解读|Nat Commun(16.6):CSTF2介导的mRNA N 6 -甲基腺苷修饰驱动胰腺导管腺癌m 6 A亚型
✦ +
+
论文ID
原名:CSTF2 mediated mRNA N6-methyladenosine modification drives pancreatic ductal adenocarcinoma m6A subtypes
译名:CSTF2介导的mRNA N 6 -甲基腺苷修饰驱动胰腺导管腺癌m 6 A亚型
期刊:Nature Communications
影响因子:16.6
发表时间:2023.10.10
DOI号:10.1038/s41467-023-41861-y
背 景
胰腺导管腺癌 (PDAC) 是世界上第四大癌症相关死亡原因,通常在晚期才诊断出来。由于缺乏有效的早期诊断、治疗方法以及治疗药物难以到达肿瘤部位,PDAC 的治疗效果并不好。基因转录本的N6 -甲基腺苷 (m6A) 修饰在癌症中发挥着关键作用。研究者团队假设 RNA 中的 m6A 可能有望作为 PDAC 亚型分型的分子标记。
实验设计
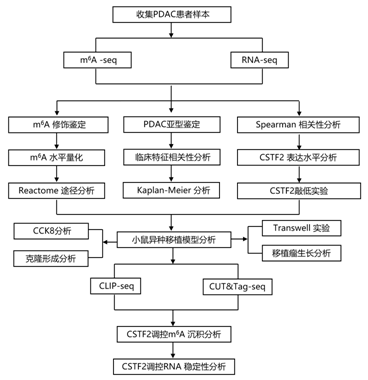
结 果
01

转录组分析m6A在PDAC中的定位
研究者团队对来自65个个体的98个胰腺样本进行了m6A测序(m6A-seq),其中包括33对PDAC和相应的正常组织以及另外32个PDAC样本,并通过MACS2和MeTPeak鉴定了17996个m6A峰(图1a)。此外,鉴定出的m6A位点富集于经典GGACH基序(图1b)和起始密码子和终止密码子附近的区域(图1c)。
在这17996个m6A位点中,与33个配对的正常组织相比,33个肿瘤中195个位点高甲基化,93个位点低甲基化(图1d)。
肿瘤和正常样本标记的排列分析(1000次)平均产生17个差异m6A位点,远远少于观察到的288个差异m6A位点,表明所鉴定的异常m6A位点不是随机的。随机选择的m6A突变位点中有96.6%通过MeRIP-qPCR验证,证明本项研究中的m6A-seq数据的可靠性(图1e)。
大多数差异m6A位点位于mrna内,富集在终止密码子周围和3'UTR区域。288个失调的m6A位点富含与癌症通路相关的基因,如细胞周期和上皮-间质转化(EMT)(图1f)。例如,已报道的癌基因如CENPF、WNT7B和NTSR1在肿瘤中与邻近正常组织相比,存在超m6a甲基化(图1g-h)。
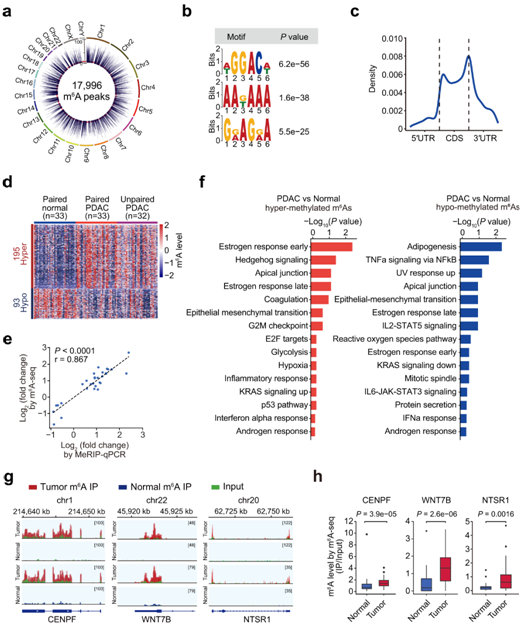
图1. PDAC 中m6A修饰的全转录组图谱。
(a) Circos图显示在 65 个 PDAC 肿瘤和 33 个正常组织样本中鉴定出的 RNA m6 A 修饰。 (b) 序列标识表示通过MEME工具分析的m6A中富集的序列基序。(c) m6A在mrna中的位置分布。 (d) 热图显示PDAC肿瘤样本中不同的m6A水平。(e) m6A-seq或MeRIP-qPCR检测29例异常m6A(肿瘤/正常)差异倍数的Spearman相关性。 (f) 通过Reactome通路分析,显著富集了高甲基化m6A(左)或低甲基化m6A(右)的特征。(g) m6 A-seq表示的肿瘤组织和邻近正常组织中 RNA中 m6 A的丰度。 (h) 肿瘤组织和邻近正常组织中m6A-seq显示的rna的丰度。
随后。他们根据这些m6A差异峰对PDAC患者进行无监督的共识聚类,进一步表征了两种PDAC亚型(S1和S2)(图2a)。S2 PDAC表现出与S1 PDAC不同的m6A模式(图2b),但在两种PDAC亚型的邻近正常组织中没有差异,这表明亚型模式是肿瘤特异性的。此外,S1和S2 PDAC样本之间差异甲基化的m6A在S1 PDAC样本与邻近正常组织样本之间没有差异(图2b),并且与邻近正常组织和肿瘤组织之间差异甲基化的m6A有很大的重叠(图2c),表明S2 PDAC特异性m6A失调。
他们分析了这两种亚型与已知临床因素的相关性,如性别、年龄、吸烟状况、饮酒状况、肿瘤分期、分化、血管侵犯和淋巴结转移。除神经浸润外,其余结果均为阴性(图2d)。用于m6A-seq的两种亚型肿瘤组织间质含量无显著差异(图2d),表明这些亚型模式是PDAC的内在特征。他们发现S2 PDAC中Bailey 's鳞状亚型和Collisson 's经典亚型的频率明显高于S1 PDAC(图2d)。
生存分析显示,S2 PDAC的中位无进展生存期(PFS)时间和总生存期(OS)时间明显短于S1 PDAC(图2e)。
图2. 通过全转录组 m6 A 修饰对 PDAC 进行亚型分析。
(a) 通过差异 m6A将 PDAC 区分为 S1 或 S2 亚型。(b) 热图显示正常、S1 和 S2 PDAC 组织中的m6 A的显著差异。 (c) 维恩图显示S2 PDAC亚型与S1 PDAC亚型的高甲基化(上)或低甲基化m6A(下)之间以及PDAC肿瘤与正常之间存在很大的重叠。 (d) m6a定义的PDAC亚型与临床(性别、年龄、吸烟状况、饮酒状况、肿瘤分期、分化、神经侵袭、血管侵袭和淋巴结转移)或分子特征(转录亚型)之间的关系。 (e) 通过Kaplan-Meier曲线分析不同m6A亚型PDAC患者的无进展生存期(PFS)(上)和总生存期(OS)(下)。
02
CSTF2促进 PDAC m6A 亚型形成
接下来他们探讨了 PDAC 亚型形成的机制。首先,发现裂解刺激因子2(CSTF2)RNA水平与S2 PDAC中高甲基化m6A位点的水平最具显著相关性(图3a)。PDAC中的CSTF2 RNA和蛋白质水平均显著高于邻近正常组织,并且S2 PDAC中的CSTF2 RNA和蛋白质水平均显著高于S1 PDAC(图3b)。
随后他们选择具有中等CSTF2表达水平的PDAC细胞系(PANC-1和SW1990)进行实验。当敲低PANC-1和SW1990细胞中的CSTF2时,差异m6A位点的甲基化水平分别显著降低86%和88%(图3c-d)。
此外,当CSTF2在相同的细胞系中异位过表达时,8804和8554个m6A位点发生超甲基化(图3e-f),在CSTF2敲低的两种细胞类型中,72.8%和61.7%的低甲基化m6A重叠(图3g)。通过MeRIP-qPCR进一步验证cstf2敲低后m6A显著失调(图3h-i)。综上所述,这些结果表明CSTF2可能调节PDAC中mRNA m6A的形成。
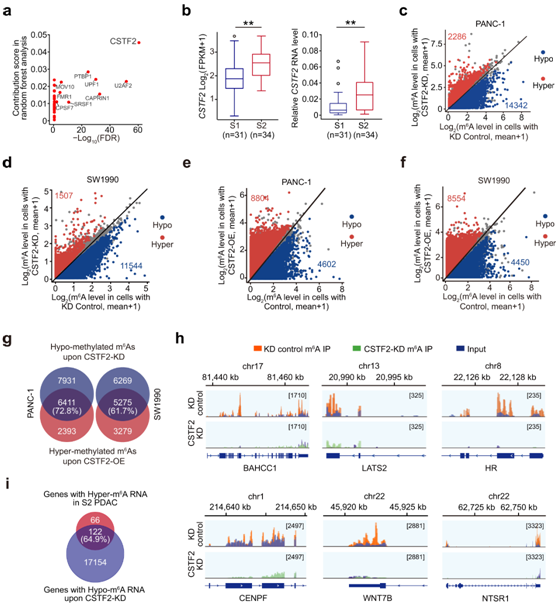
图3. CSTF2 是促进S2 PDAC 亚型中mRNA m6A 沉积的关键蛋白。
(a) S2 PDAC 亚型中RNA 结合蛋白 (RBP) 和高甲基化 m6 A表达之间的 Spearman 相关性。(b) 通过 RNA-seq(左)或 qRT-PCR(右)确定两种 PDAC 亚型中的 CSTF2 表达水平。(c) CSTF2对PANC-1细胞中m6A水平的影响。 (d) CSTF2对SW1990细胞中m6A水平的影响。(e-f) CSTF2强制表达或不表达时PANC-1细胞和SW1990细胞 m6A水平的散点图。 (g) 维恩图显示了CSTF2敲低细胞中低甲基化的m6A和CSTF2过表达细胞中高甲基化的m6A之间的重叠。(h) 整合基因组浏览器(IGV)显示,在具有或不具有CSTF2 KD的PDAC细胞中,转录本中m6A的丰度不同。(i) 维恩图显示S2 PDAC 亚型中具有高 m6A的 mRNA 和具有CSTF2 KD的 PDAC 细胞中具有低 m 6A 的 mRNA 。
03
CSTF2促进PDAC细胞的恶性表型
然后他们探讨了 CSTF2 对 PDAC 细胞恶性表型的影响。体外实验表明,CSTF2的敲低显著抑制了PDAC细胞的细胞增殖、克隆形成、细胞周期、迁移和侵袭的能力(图 4a-d)。通过使用小鼠皮下异种移植模型,他们还发现CSTF2沉默会显著抑制PDAC肿瘤的生长速率(图4e)。此外,CSTF2的强制表达促进了PDAC细胞的肺转移,而CSTF2敲低则显示出相反的效果(图 4f)。
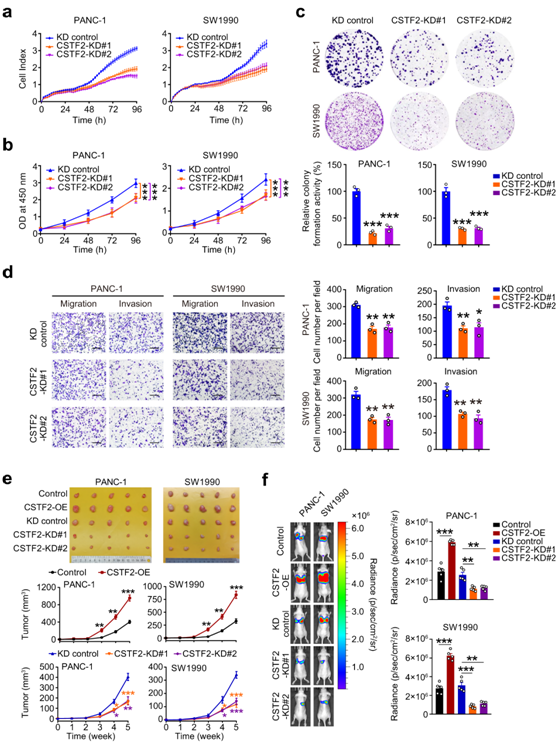
图4. CSTF2的敲低抑制PDAC细胞的增殖和转移。
(a-b) CSTF2 KD 抑制 PDAC 细胞增殖。(c) CSTF2 KD 抑制 PDAC 细胞克隆形成。 (d) 通过 Transwell 实验测定,CSTF2 KD 抑制 PDAC 细胞迁移和侵袭。(e) CSTF2对裸鼠体内PDAC细胞异种移植肿瘤生长的影响。(f) 通过尾静脉注射细胞,CSTF2对裸鼠中 PDAC 细胞肺定位的影响。
04
CSTF2通过延迟伸长介导 m6A 沉积
他们的CLIP测序数据显示CSTF2 RNA结合位点与RNA中的m6A位点有很好的重叠(图5a-b)。先前的研究报道,CSTF2可以直接与RNA聚合酶II (RNA Pol II)相互作用,而RNA聚合酶II已知通过共转录募集m6A甲基转移酶复合物(MTC),这表明CSTF2可能通过MTC和RNA Pol II影响m6A的沉积。因此,他们对CSTF2和RNA Pol II进行了CUT&Tag测序,结果显示CSTF2和RNA Pol II的基因组结合位置有很好的重叠(图5c)。与没有Pol II占据的峰相比,CSTF2敲除后,Pol II占据的m6A峰值的m6A水平明显降低(图5d)。此外,他们发现在Pol II占据中表现出显著变化的基因在CSTF2敲低后也表现出m6A水平的更大降低(图 5e)。此外,他们发现DNA中的CSTF2结合位点与RNA中的CSTF2结合位点和m6A位点共定位,并且该共定位与RNA Pol II相关(图5f-g)。这些结果表明 RNA Pol II 可能确实在介导 CSTF2 调节的 m6A 沉积中发挥作用。
他们进一步对CSTF2敲低后的 Pol II 和 Pol II-Ser2P 进行了 CUT&Tag 测序。CSTF2敲低后,在具有低甲基化-m 6 A的基因中观察到 RNA Pol II 和 Pol II-Ser2P 密度显著降低,而 CSTF2 非靶标的 RNA Pol II 密度不受影响(图 5h)。他们还观察到 H3K79me2 和 H3K36me3 略有增加,但 Pol II-ser5P 的富集没有显著变化(图5i)
他们还发现CSTF2敲低促进了PDAC细胞中新生RNA的合成,但CSTF2的异位过度表达减弱了新生RNA的合成(图5j-k),证实CSTF2作用降低了Pol II的延伸率。他们发现PDAC细胞中CSTF2表达的强制变化导致RNA Pol II和METTL3相互作用的显著变化(图 5l-m)。值得注意的是,敲除CSTF2引起的整体m6A水平下降与敲除METTL3引起的m6A水平下降相当,并且敲除METTL3的细胞中低甲基化的m6A与CSTF2产生的69%的m6A重叠(图5n)。CSTF2敲低导致METTL3在目标转录本m6A区域周围的结合减少,但CSTF2的异位过表达增强了相互作用(图5o-p)。
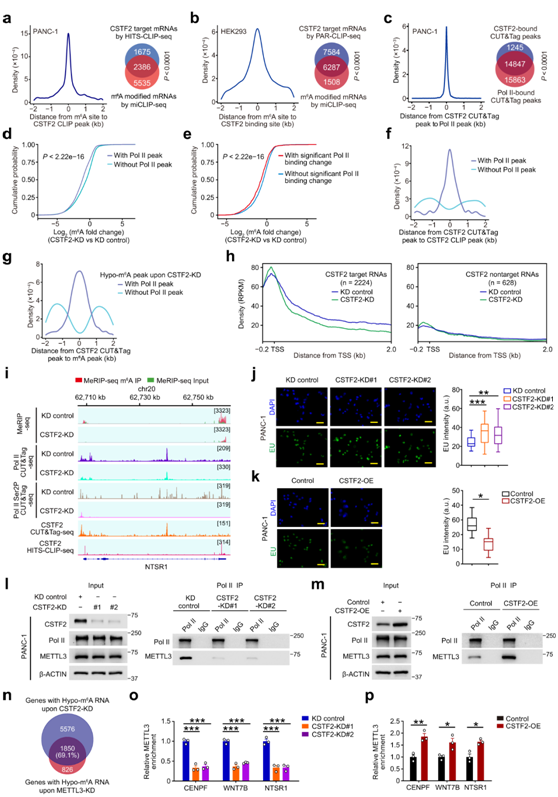
图5. CSTF2通过延缓延伸介导m6A沉积。
(a-b) PANC-1和HEK293 细胞中 RNA 中 CSTF2 结合位点和 m6A 位点的共定位。(c) PANC-1细胞中CSTF2和RNA Pol II的DNA结合位点的共定位。(d-e) 富含Pol II结合的基因(d)或Pol II结合变化显著的基因(e)在CSTF2-KD上的m6A变化更为剧烈。(f) PANC-1 细胞中 CSTF2 的 DNA 和 RNA 结合位点的共定位。(g) PANC-1 细胞中 CSTF2 的 DNA 结合位点和 RNA m6A 位点的共定位。 (h) CSTF2-KD后PANC -1细胞中沿CSTF2靶mRNA的RNA Pol II密度的比较。(i)显示的是在CSTF2 KD上经历 m6A 水平和 Pol II 结合密度变化的转录本的代表性轨迹。(j-k) 5-乙基尿嘧啶(EU) 标记实验。(l-m) 显示CSTF2 KD或过表达对PANC-1细胞中Pol II和METTL3相互作用的影响。(n) 显示了在CSTF2 KD和METTL3 KD上具有亚m6a的mrna之间的重叠。(o-p) CLIP-qPCR显示,CSTF2 KD在PANC-1中受损,但过表达增强METTL3与靶转录物的结合。
05
CSTF2 调节的 m6A 增强 RNA 稳定性
然后,他们探究了 m 6 A对 PDAC 中宿主 RNA 水平的影响,发现S1 和 S2 PDAC 亚型之间的205个差异甲基化 m6A(148个RNA)对其宿主 RNA 水平有影响(图6a)。在148个RNA中,与S1 PDAC相比,S2 PDAC中115个RNA的m6A水平和RNA水平均上调,而33个RNA的m 6A水平和RNA水平均下调(图 6a)。在PDAC细胞中也观察到m6A水平与RNA水平的正相关,CSTF2敲低后,许多RNA的水平下调会导致m6A发生低甲基化,但只有少数RNA会导致3'UTR的延长,这表明CSTF2调节的m6A而不是CSTF2调节的可变聚腺苷酸化(APA)可能有助于RNA水平的升高。
此外,他们进行了基于dCas13的m6A编辑和gRNA特异性操纵m6A位点(图6c),证实了m6A水平下调,抑制了IGF2BP2的结合,导致mRNA水和转录本半衰期下降(图6d-g)。强制表达IGF2BP2无法挽救m6A水平下调对mRNA水平和转录本半衰期的影响(图6h-i)。综上所述,cstf2调控的m6A通过IGF2BP2增强了RNA的稳定性。
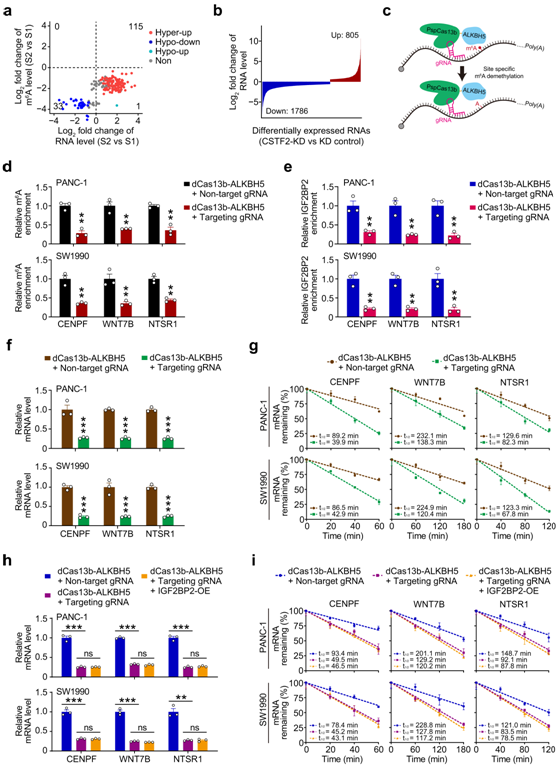
图6. 异常的 m6A 增强 mRNA 稳定性。
(a) m6A水平与其宿主 RNA 水平的相关性。 (b) PANC-1细胞中CSTF2敲低后差异表达的CSTF2靶RNA的瀑布图。(c) 基于dCas13的m6A编辑系统框图。(d) 通过 MeRIP-qPCR 检测的CENPF、WNT7B、NTSR1转录本的相对 m6A 富集。 (e) 通过 CLIP-qPCR 检测转录本的相对 IGF2BP2 富集。(f-i) 通过 qRT-PCR 检测到的CENPF、WNT7B、NTSR1的相对 mRNA 水平和半衰期。
+ + + + + + + + + + +
结 论
本项研究对来自 65 名PDAC患者的 98 个组织样本进行m6A-seq。与邻近正常组织相比,在 PDAC 中鉴定出 17996 m6A 峰,其中 195 个高甲基化和 93 个低甲基化。差异性 m6A 修饰区分了具有不同预后结果的两种 PDAC 亚型。这两种亚型的形成是由新发现的 m6A调节因子 CSTF2 驱动的,CSTF2 通过减慢基因转录过程中 RNA Pol II 的延伸率来调节 m6A甲基化。大多数CSTF2调节的m6A对宿主基因的RNA水平具有积极影响。这些结果为 PDAC 的精准医疗提供了一种有前景的 PDAC 分型策略和新的理论依据。
+ + + + +



