

 English
English文献解读|Gut Microbes(12.2):多组学分析揭示阿片类药物使用后肠道微生物组与宿主之间的相互作用
✦ +
+
论文ID
原名: Multi-omics analysis revealing the interplay between gut microbiome and the hostfollowing opioid use
译名:多组学分析揭示阿片类药物使用后肠道微生物组与宿主之间的相互作用
期刊:Gut Microbes
影响因子:12.2
发表时间:2023.08.23
DOI 号:10.1080/19490976.2023.2246184
背 景
过去几十年来,阿片类(Opioid)药物危机在美国一直存在。阿片类药物使用相关的微生物失调正在成为肠道稳态和阿片类药物行为反应的关键调节因子。然而,目前尚不清楚微生物群落在调节宿主反应中的作用的机制。
实验设计
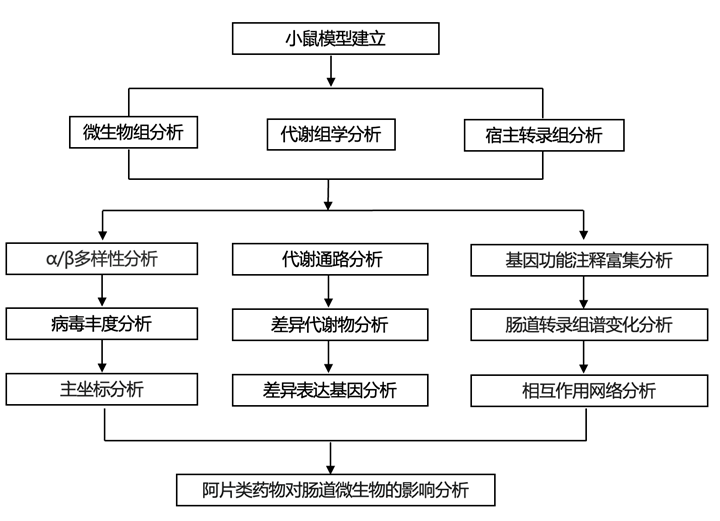
结 果
01

吗啡治疗诱导小肠微生物组的分类和功能失调
为了了解吗啡治疗对肠道微生物组的影响,研究团队对小鼠回肠微生物组进行了全基因组测序。他们在物种层面总共鉴定了 63 个分类群(47 个细菌、3 个真菌、1 个古细菌、11 个病毒和 2 个噬菌体)。β多样性分析(图1a)根据组合分类群的 Bray-Curtis 相异矩阵计算得出,显示了对照组和治疗组的明显聚类。LEfSe 识别出的显著改变的类群,其p值 < 0.05 且 LDA 得分 > 2.0 在分支图中表示(图1b)。对细菌分类群的进一步分析揭示了与对照小鼠相比,吗啡治疗小鼠的α多样性(香农指数和辛普森指数)和β多样性(Bray-Curtis)发生了变化。吗啡处理导致Parasutterella excrementihominis、Burkholderiales bacter 1_1_47、粪肠球菌、Staphylococus xylosus、Firmicutes bacter M10-2、Bifidobacterse pselongum和Enterorhabdus caecimuris显著扩增,并消除了约氏乳杆菌(Lactobacillus johnsonii)(图1b)。
接下来,他们分析了宏基因组数据,通过使用 HUMAnN 管道映射微生物读数来识别功能变化。吗啡治疗导致 201 个 KEGG基因 (KO)发生显著改变。基于对照组和吗啡处理组中 KO 相对丰度的主坐标分析 (PCoA) 图表明,吗啡显著改变了细菌群落的功能特征(图1c)。此外,代谢通路分析表明吗啡治疗增加了氨基酸转运和代谢、辅因子和维生素代谢、脂质代谢,并减少外源生物降解和代谢以及碳水化合物代谢(图1d)。在吗啡治疗小鼠的宏基因组中检测到较高丰度的通路参与细菌细胞膜成分的产生和运输,包括脂磷壁酸(LTA)生物合成过程、肽聚糖周转、革兰氏阴性细菌细胞外膜组装、脂多糖(LPS)结合和运输(图1e)。
他们还观察到,与对照组相比,吗啡处理组中参与脂质代谢的通路富集,包括脂质A和磷脂酰甘油生物合成过程,磷脂转运和脂蛋白转运活性,而参与脂肪酸代谢过程和脂质生物合成过程的通路下调(图1f),表明吗啡处理组的微生物群中存在脂质代谢的失调。此外,与对照组相比,吗啡微生物组中涉及维生素(维生素B1、B2、B5和叶酸)的生物合成(图1G)和氨基酸(包括谷氨酸)的生物合成以及色氨酸分解为犬尿氨酸的通路显著富集(图1h)。此外,在吗啡处理的小鼠的微生物组中,观察到参与病毒防御反应的通路丰度增加,并且参与吗啡6-脱氢酶活性、从宿主细胞释放病毒和溶菌酶活性的通路减少(图1i)。这些结果表明,吗啡的使用导致了多界水平的成分改变,也诱导了细菌的功能失调。
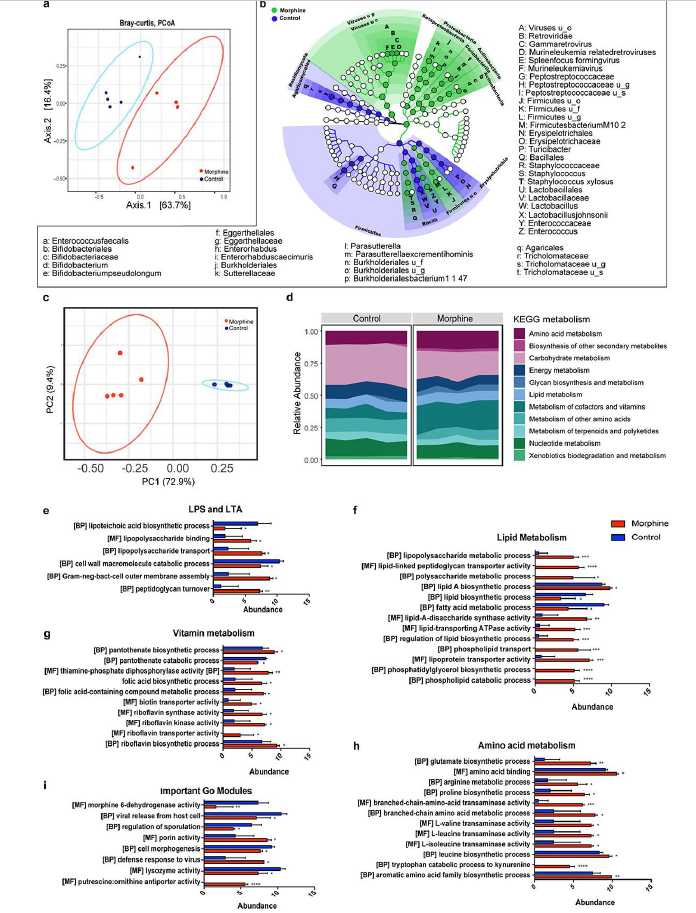
图1. 吗啡治疗会导致小肠微生物组的分类和功能失调。
(a) 对照和吗啡组中物种水平的回肠微生物组系统发育相对丰度的β多样性。(b) 分支图显示了差异丰度的微生物类群。 (c) 基于 KO 相对丰度的主成分分析(PCA)区分对照和吗啡微生物组的功能特征。 (d) 基于对照组和吗啡组中 KEGG 代谢的微生物组功能概况。(e-i) GO分析。
02
吗啡的使用会导致小肠代谢组发生广泛变化,并突出脂质代谢的变化
为了充分表征成分和功能失调的影响,他们使用非靶向液相色谱-质谱 (LC-MS) 对吗啡治疗和对照小鼠的回肠腔内容物进行了代谢物分析,鉴定并注释了 243 种代谢物,其中 115 种代谢物(53 种耗竭,62 种富集)因吗啡治疗而发生显著改变。偏最小二乘判别分析 (PLS_DA) 图将吗啡代谢组与对照组区分开来 (图2a)。吗啡治疗后的代谢变化如火山图所示(图2b),前 50 个改变的代谢物以热图表示(图2c)。各种脂质代谢物的富集包括乙醇胺[N-油酸乙醇胺(OEA)、亚油酸乙醇酰胺(LEA)、α -亚麻烯基乙醇酰胺(αLEA)和棕榈酰乙醇酰胺(PEA)],磷酸胆碱(PC)[PC(O-20:1)、 PC(13:0/18:3 (9z,12z,15z)、PC(35:2)、PC(0-18:0)、 PC(35:3)]、脂肪酸(FA)[肉豆豆酸、甲基花生酸、FA(22:0)、FA(20:2)、FA(22:4)]、丙酰肉碱、l -己酰肉碱、十二酰肉碱和亚油酸的代谢物(图2c-f)。吗啡组维生素核黄素、黄酮类(染料木黄酮、大豆苷、柚皮素1-o-葡萄糖苷、汉黄芩苷、黄芩苷)、左卡尼汀、亚精胺和胆汁酸(熊去氧胆酸、α -鼠胆酸、ω -鼠胆酸、胆酸、猪胆酸、甘胆酸)的消耗明显减少(图2c-f)。他们观察到吗啡代谢物吗啡-6-葡萄糖苷酸 (M6G) 显著富集,表明吗啡的代谢发生了改变(图2c)。
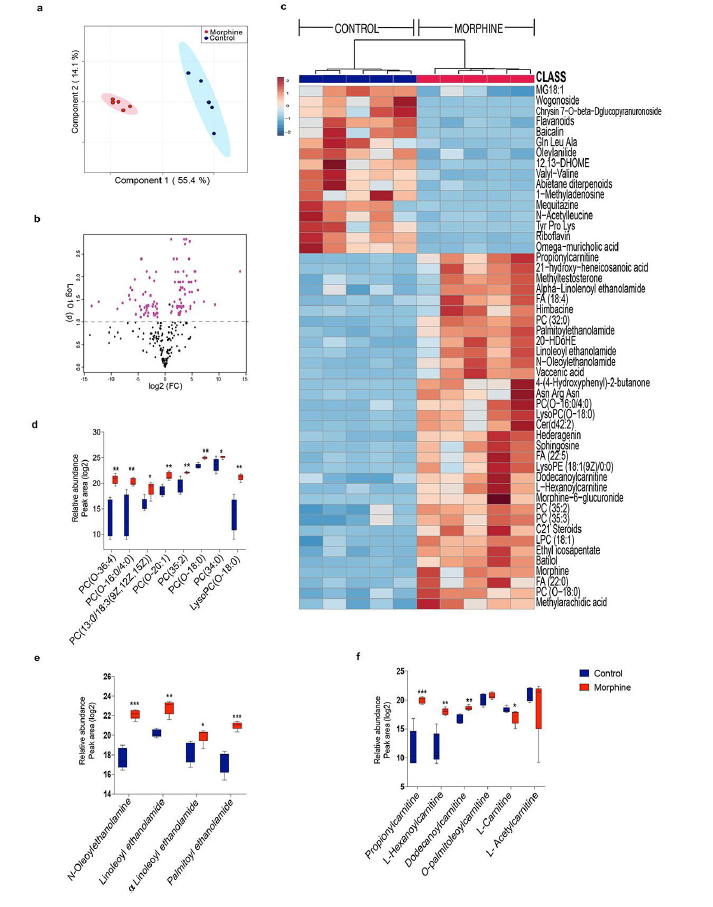
图2. 吗啡的使用会导致小肠代谢组发生广泛变化。
(a) 显示吗啡和对照回肠代谢物谱的偏最小二乘判别分析 (PLS-DA) 。(b) 火山图显示通过单变量分析确定的最重要的代谢物。 (c) 热图显示对照组和吗啡组中前 50 种代谢物发生显著变化。 (d-f) 箱线图显示磷酸胆碱、N-酰基乙醇胺和肉碱的相对丰度。
03
吗啡诱导的小肠转录组变化包括炎症相关基因的表达增强和脂质代谢的改变
他们定义了由 5140 个(2606 个上调,2534 个下调)差异表达基因 (DEG) 组成的核心回肠吗啡基因表达特征,并利用ClueGo和ToppCluster进行基因富集分析。总体而言,上调的DEG的ClueGo饼图显示免疫细胞趋化性和相关的体液和细胞因子反应显著富集(图3b)。同时,还观察到针对原生动物和细菌分子的富集反应(图3b)。他们观察到总体代谢过程急剧减少(图3c)。对上调基因和下调基因进行ToppCluster分析,详细描述了富集的生物过程,包括重要通路(图3d-e)。与对照相比,吗啡上调的基因特征显示出与防御反应相关的通路的富集,例如白细胞介素信号传导通路、先天性和适应性免疫细胞激活、对细菌和 LPS 的反应等。参与多种代谢过程(包括类固醇、脂质、酒精生物合成)的基因下调(图3e)。他们还注意到参与脂质结合、脂质转运和脂质分解代谢过程的通路上调,表明吗啡治疗后代谢过程普遍失调。
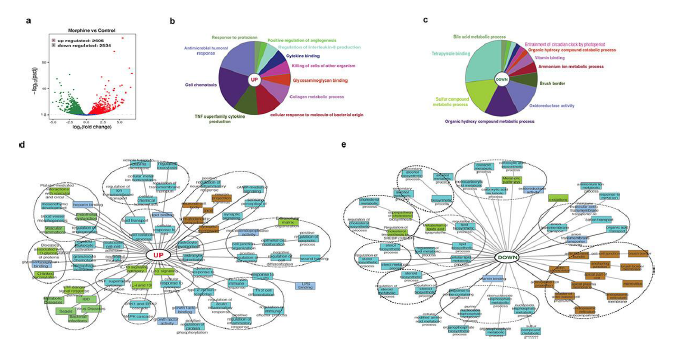
图3. 吗啡治疗小鼠小肠转录组变化的总体概述。
(a)吗啡组和对照组之间 5140 个差异表达基因的火山图。(b-c) 使用 ClueGO 图对吗啡处理后2606 个上调基因和2534 个下调基因进行功能注释富集分析。 (d-e) 详细的功能注释富集分析。
04
吗啡介导的小鼠肠道转录组谱的变化受到肠道微生物群的影响
为了深入了解吗啡治疗的失调微生物组特征对宿主转录组的全基因组影响,他们比较了吗啡治疗组和在没有微生物组的情况下用吗啡治疗的 Abx+吗啡 (AM) 组的 mRNA 表达水平。与 AM 组相比,他们在吗啡治疗组中观察到 1371 个 DEG(667 个上调和 704 个下调),表明1371个基因的表达是由吗啡微生物组介导的(图4a)。ClueGo 饼图描绘了各种免疫调节过程的富集以及细胞对生物刺激的反应,例如吗啡组中的细菌、病毒或真菌来源的分子(图4b)。吗啡治疗后还观察到对异生物质刺激的反应同时下降(图4c)。使用 ToppCluster 分析对 DEG 进行进一步分析显示,与 AM 组相比,吗啡组的先天性和适应性免疫反应以及细胞因子信号传导有所增加,这表明菌群失调是炎症的介质(图4d)。
在吗啡组中,包括氨基酸代谢过程、酒精代谢过程、脂质和脂蛋白代谢过程在内的多种代谢过程通路发生大幅下调(图4e),表明微生物组在这些代谢过程中的作用。与肠通透性相关的细胞成分如紧密连接、细胞-细胞连接、锚定连接、肌动蛋白细胞骨架等也出现显著下调(图4e)。综上所述,对吗啡组和 AM 组基因表达模式的比较分析发现,生态失调的微生物群是炎症和代谢失调的介质。

图4. 吗啡介导的小鼠肠道转录组谱变化受到肠道微生物群的影响。
(a) 吗啡组和 AM 组之间 1371 个差异表达基因的火山图。(b-c) 基因功能注释富集分析。 (d-e) 详细功能注释富集分析。
05
整合吗啡介导的微生物组、代谢组和宿主转录组变化的多组间系统分子相关性
接下来,为了确定微生物与宿主的直接相互作用和代谢物介导的间接相互作用,他们构建了一个涵盖三个组学测量的大规模相互作用网络。利用 Spearman 相关性,确定了其表达与微生物组变化共变的代谢物和 DEG,还包括随代谢物变化而共变的各种 DEG。他们确定了三个数据集之间的 15617 个显著相关性。通过整合类群、代谢物和 DEG 之间的显著Spearman 相关性,生成用于可视化的网络(图5)。
细菌P. excrementihominis和B. bacterium 1_1_47在网络中连接较好,占相互作用的数量较多,分别关联76%和75%的节点。P. excrementihominis和B. bacterium 1_1_47与M6G、乙醇胺、脂肪酸、磷酸胆碱呈正相关,并与许多参与炎症和免疫反应的基因存在显著正相关(图5)。致病菌分类群taxa E. faecalis 和S. xylosus与长链脂肪酸代谢产物肉豆蔻烯酸、三糖酸、甲基花生四烯酸、二十碳二烯酸和二十二糖酸呈正相关。粪肠杆菌也与乙醇胺的变化有关。E. faecalis 、S. xylosus和LCFA与参与脂质结合、转运、定位和分解代谢过程的基因相关,暗示这些类群与脂质代谢功能障碍有关。N-酰基乙醇胺(NAE)水平升高与约氏乳杆菌和消化链球菌呈负相关,与P. excrementihominis、B. bacterium 1_1_47, E. faecalis、E. caecimuris和 A. municiphila.呈强正相关。
益生菌约氏乳杆菌和共生的未分类的消化性链球菌科的负相关数量最多,分别占负交互作用的44%和9%。他们还观察到益生菌约氏乳杆菌与几种肠道代谢物,包括 PC (32:0)、PC (0-18:0)、PC(35:2)、黄酮(batilol和hederagenin)、乙醇胺和FA(20:4)呈强负相关(图5)。约氏乳杆菌与参与脂质代谢和脂质生物合成通路的基因呈正相关,包括脂肪酸结合蛋白6 (Fabp6) 、酰基辅酶 A 合成酶家族成员 2 (Acsf2)以及参与细胞连接的基因如Abcc2、Ank3、Pmp22、Mmp7、Mmp9、Mmp10、Mmp19等(图5)。此外,约氏乳杆菌和消化链球菌科与涉及免疫细胞趋化性和对细胞因子刺激反应的相关基因呈负相关。
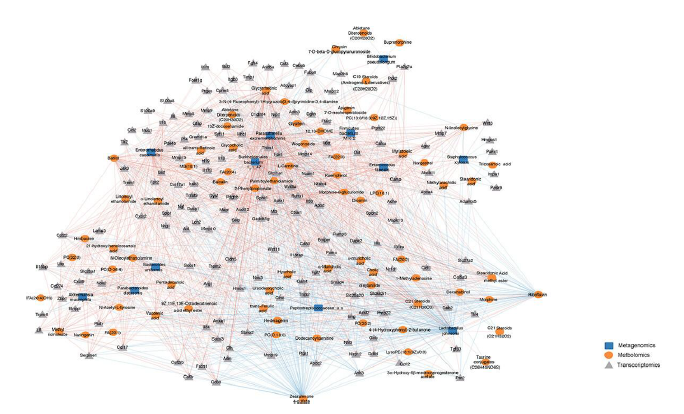
图5. 整合吗啡介导的微生物组、代谢组和宿主转录组变化的多组间系统分子相关性。
+ + + + + + + + + + +
结 论
本项研究利用全基因组测序、非靶向代谢组学和 mRNA 测序来分别识别微生物组、代谢组和宿主转录组的变化。吗啡处理导致Parasuterella excrementihominis、Burkholderiales bacterium 1_1_47、Enterococcus faecalis、Enterorhabdus caecimuris的显著扩增和约氏乳杆菌的消耗显著增加,这些变化与脂质代谢物和类黄酮的变化相关。在吗啡处理的动物的微生物组中观察到微生物代谢(脂质、氨基酸、维生素和辅因子的代谢)的显著改变以及毒力因子表达和LPS和LTA生物合成的增加。本项研究在宿主转录组中观察到先天性和适应性免疫反应、脂质代谢和肠道屏障功能障碍的广泛变化。本项研究还确定了限制微生物失调的潜在治疗干预领域,并为阿片类药物研究界提供了新的理论依据。
+ + + + +



