

 English
English文献解读|Nat Cancer(22.7):中国人群乳腺癌的综合多组学分析揭示了患者分层和治疗脆弱性
✦ +
+
论文ID
原名:Integrated multiomic profiling of breast cancer in the Chinese population reveals patient stratification and therapeutic vulnerabilities
译名:中国人群乳腺癌的综合多组学分析揭示了患者分层和治疗脆弱性
期刊:Nature Cancer
影响因子:22.7
发表时间:2024.02.12
DOI号:10.1038/s43018-024-00725-0
背 景
乳腺癌是影响女性的最常见的恶性肿瘤,这凸显了对有效治疗策略的迫切需要。分子谱分析指导乳腺癌精准治疗;然而,在公开的大规模研究中,亚洲患者的代表性不足。
实验设计
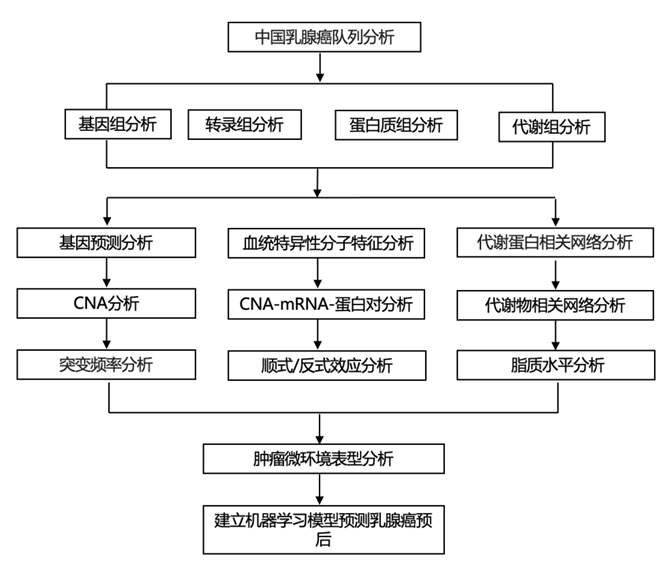
结 果
01

CBCGA 队列的多组学前景
本项研究对一个大型中国乳腺癌队列进行了多组学分析,即中国乳腺癌基因组图谱(CBCGA)项目,纳入了2013年至2014年期间在复旦大学附属肿瘤医院接受治疗的来自中国全国的773名乳腺癌患者,获得了 653 例肿瘤组织和配对血液样本的全外显子组测序 (WES) 数据,还收集了 685 个组织的拷贝数改变 (CNA) 数据、752 个组织的 RNA 测序 (RNA-seq) 数据、278 个组织的蛋白质组学数据和 453 个原发性肿瘤组织的代谢组学数据。
为了进一步分析,研究者团队对微阵列(PAM50)乳腺癌固有亚型进行了基因预测分析。在752份具有RNA-seq数据的样本中,29.5%为管状A型, 29.4%为管状B型, 19.7%为人表皮生长因子受体2(HER2)富集,14.9%为基底样型, 6.5%为正常样型(图1)。管腔A、管腔B、HER2富集、基底样和正常样的中位肿瘤负担(TMB)分别为0.62、0.91、1.32、1.29和0.65个突变/ Mb(图1a)。突变特征在PAM50亚型中的分布也各不相同。年龄相关特征(单碱基取代(SBS)1/SBS5)在管腔A、管腔B和正常样肿瘤中占主导地位。APOBEC相关特征(SBS2/SBS13)在HER2富集的肿瘤中更为常见。相比之下,基底样肿瘤具有更高比例的同源重组缺陷(HRD)相关特征(SBS3)(图1b)。CBCGA队列中最常突变的基因包括TP53(39.8%)、PIK3CA(38.4%)、GATA3(10.3%)、MAP3K1(8.2%)、KMT2C(7.0%)和AKT1(6.3%)(图1c) 。例如,TP53在75.86%的基底样肿瘤中发生改变,明显超过其他亚型。相比之下,AKT1突变在管腔A型肿瘤中比其他亚型更为普遍。对于种系突变,683名患者有可用的白细胞DNA样本。鉴定出BRCA2突变(3.2%),其次是RAD51D(1.1%)、BRCA1(0.8%)、PALB2(0.5%)和CHEK2(0.2%)突变(图1d)。
体细胞CNA的评估包括在癌症显著靶点基因组鉴定(GISTIC)峰中检测已报道的致癌基因和肿瘤抑制基因(图1e)。HER2 富集的癌症与其他 PAM50 亚型形成鲜明对比,ERBB2扩增(GISTIC 定义)发生在 65.4% 的肿瘤中。此外,他们还鉴定了PAM50亚型之间差异表达的mRNA、蛋白质和代谢物(图1f-h)。总的来说,本研究提供了中国乳腺癌的多组学数据集,实现了跨祖先的综合分析和比较
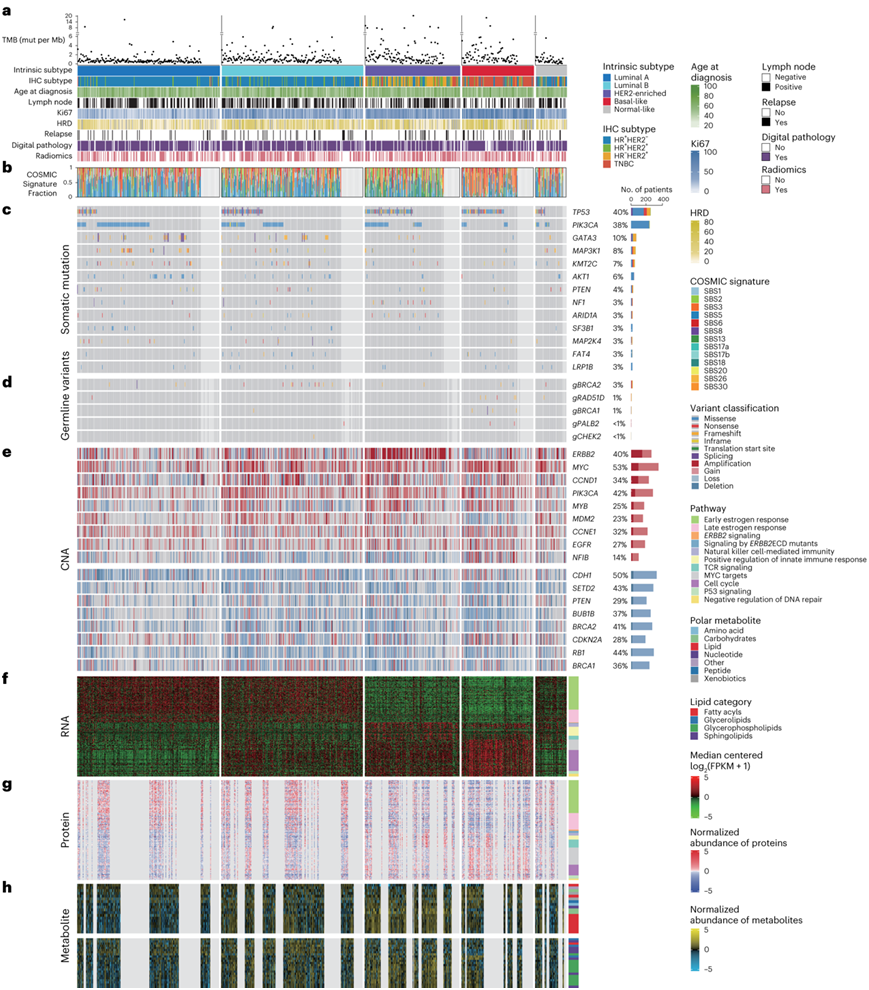
图1. CBCGA 队列的多组学景观。
(a) 总共752个乳腺癌样本,带有TMB、临床数据和分子特征注释。(b) 每个样本中癌症体细胞突变目录 (COSMIC) 突变特征的分数。(c) 体细胞突变分析。基因按 CBCGA 队列的总突变频率排序。(d) 已确定的乳腺癌易感基因中的致病性和可能致病性种系变异。 (e) 癌症相关基因的CNA。显示的癌症相关基因均位于显著的 GISTIC 峰中。 (f-h) 不同亚型之间差异表达的 mRNA蛋白质和代谢物。
02
中国患者乳腺癌的血统特异性分子特征
他们在血统特异性的背景下分析了 CBCGA 的临床和分子特征(图2)。他们重点关注诊断为浸润性导管癌 (IDC) 的病例,首先检测了这两个群体之间的基因组差异,并观察到AKT1的突变频率显著不同(图2a)。亚组分析显示,管腔 A 肿瘤在 CBCGA 中表现出AKT1突变患病率显著增加(图2b)。他们根据 Bradley-Terry 模型推断的突变获得的相对时间对体细胞改变进行排序(图2c)。AKT1突变是管腔A型IDC 病例中的早期事件,强调了其在管腔A 型乳腺癌肿瘤发生中的关键作用。
然后他们研究了 CBCGA 和 TCGA 白人个体群体之间分子亚型的差异(图2d)。与 TCGA 白人个体相比,CBCGA 队列具有明显的内在亚型分布,管腔 A 亚型比例下降,而 HER2 富集亚型比例增加。具体而言,CBCGA队列中39.4%的HR HER2样本为HER2富集亚型,而TCGA白人中只有15.3%的HR+ HER2+样本为HER2富集亚型。相应地,ERBB2在CBCGA队列中更常扩增,特别是在HR+ HER2+亚型中(图2e-f)。在CBCGA队列的HR HER2亚型中,ERBB2的高扩增与下游mRNA和蛋白的表达显著相关(图2g)。该综合分析揭示了中国队列中HER2富集程度更高的亚型,特别是在HR+ HER2+亚型中,这表明抗HER2治疗在该亚组中更大的患者队列中具有治疗益处。
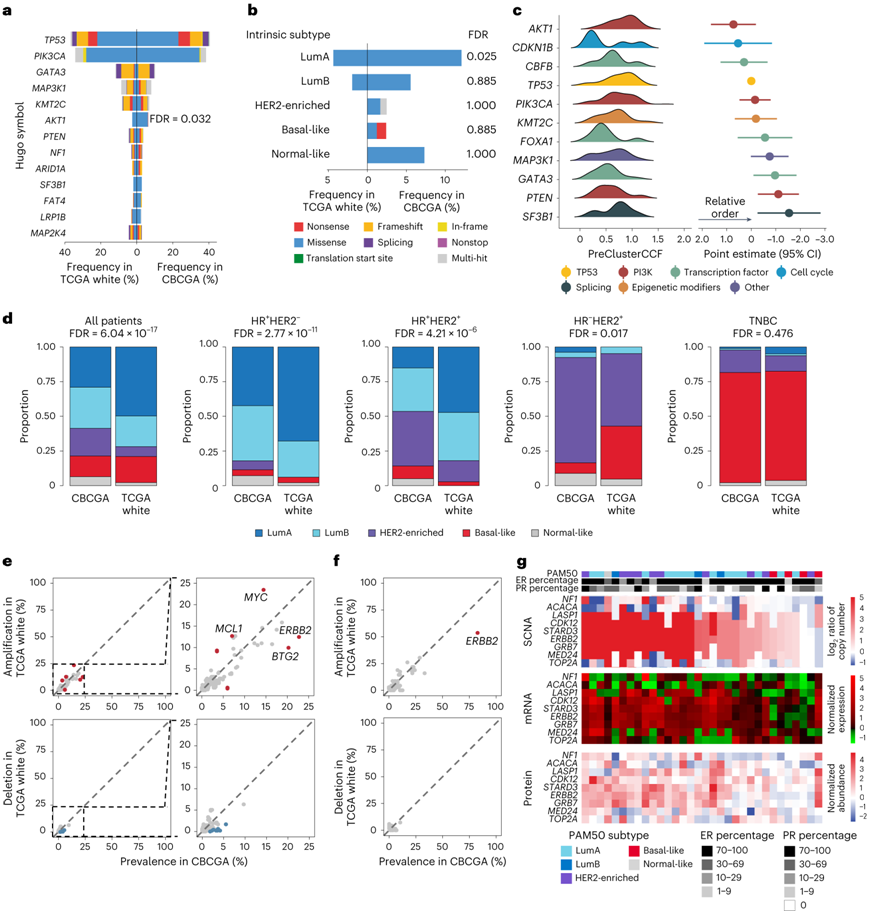
图2. 中国患者乳腺癌的血统特异性分子特征。
(a) CBCGA 和 TCGA 白人队列中 IDC 病例的基因水平突变频率。 (b) 比较CBCGA和TCGA白种人不同亚型IDC病例中AKT1突变频率。 (c) CBCGA LumA亚型突变获得的相对顺序。(d) CBCGA和TCGA白人组IDC病例的PAM50亚型。(e-f) 分别是所有亚型和 HR+HER2+亚型中 CBCGA 和 TCGA 白人个体队列之间 IDC 病例的 CNA。 (g) HR + HER2 +亚型中 17q 染色体上靠近ERBB2的基因扩增,随后在 mRNA 和蛋白质水平上相关基因过表达 。
03
蛋白质基因组分析为乳腺癌亚型提供了新的见解
CNA 在乳腺癌中很重要,可以充当“驱动力”。他们研究了 CNA 对 mRNA 和蛋白质水平的影响。通过基于同量异位串联质量标签 (TMT) 标记的技术,主要鉴定了 9787 个独特的蛋白质。在整个队列中,在5672(76.7%)个CNA-mRNA和3326个CNA-mRNA蛋白质对中观察到显著的顺式效应,而在转录和翻译水平分别在6235和3717个基因中发现了反式效应(图3a-b)。基于庞大的蛋白质基因组队列,他们随后研究了不同内在亚型的CNA的后果(图3c)。在管腔A、管腔B、HER2富集型和基底样亚型中,分别有1925、4219、2574和3426对CNA-mRNA对呈顺式表达。在这些CNA-mRNA对中鉴定的基因中,大多数也表现出一致的CNA-蛋白顺式效应。管腔B 肿瘤在蛋白质水平上表现出最高水平的一致性,其次是管腔A、基底样肿瘤和 HER2 富集肿瘤。在正常乳腺肿瘤中,只有 13 个基因在蛋白质水平上表现出显著的顺式效应。
为了探索潜在的亚型特异性驱动因素和可靶向的CNA事件,他们重点关注位于焦点拷贝数扩增峰并在至少一种亚型中的蛋白质和mRNA水平上显示顺式效应的基因(图3d)。ERBB2(位于 17q12)扩增在管腔B 和 HER2 富集肿瘤中均显示出显著的 CNA-mRNA-蛋白质顺式效应,其中 HER2 富集亚型表现出最高的扩增频率,这与ERBB2在 HER2 富集肿瘤中的关键作用一致。
为了充分利用蛋白质组学数据,他们整合了mRNA和蛋白质丰度来细化患者分类,并利用相似性网络融合(SNF)发现了4个乳腺癌聚类(图3e)。与PAM50固有亚型相比,综合聚类能更好地对整体人群的预后进行分层(图3f-i)。
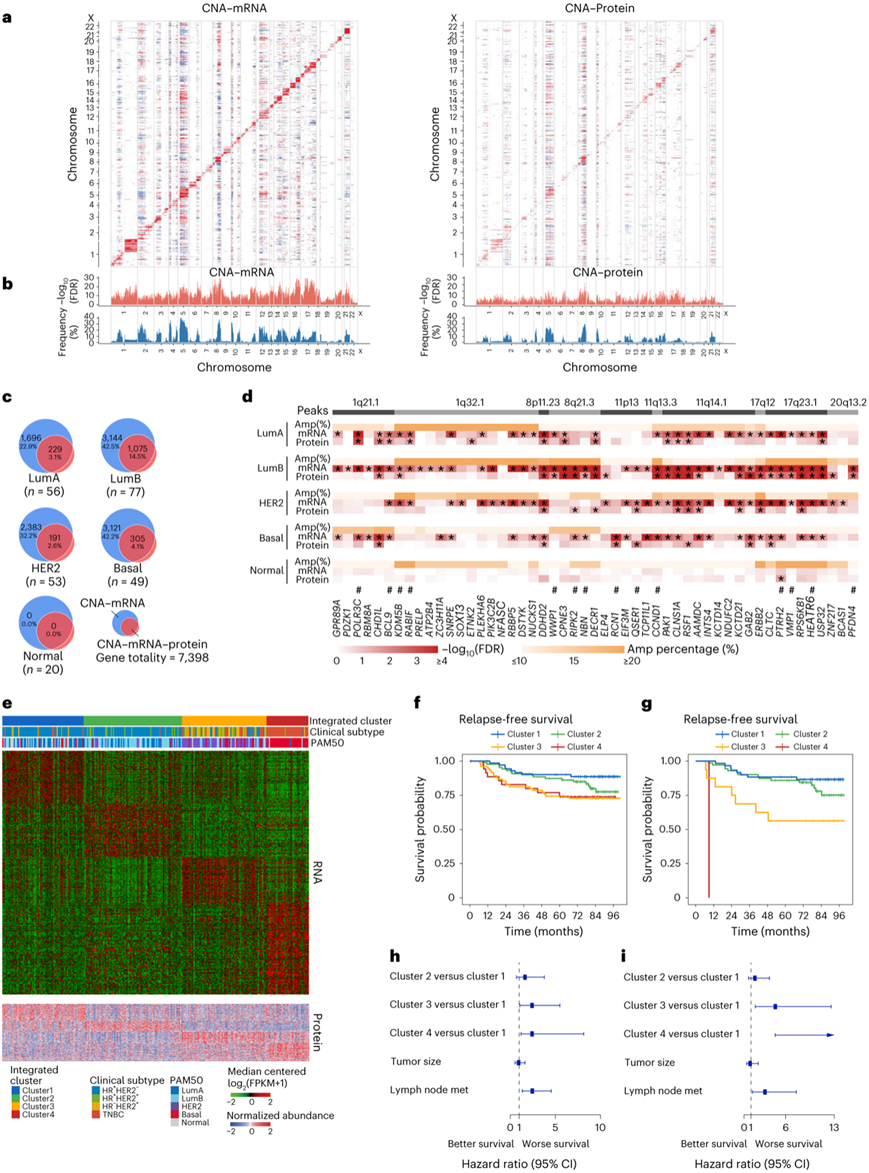
图3. 蛋白质基因组分析为乳腺癌亚型提供了新的见解。
(a) CNA 和 mRNA 表达(左)以及蛋白质丰度(右)之间的相关图。 (b) 直方图显示CNA 对 mRNA 和蛋白质的顺式和反式作用。 (c) 维恩图描绘了CNA 沿着 PAM50 亚型中的中心法则的顺式效应。(d) 位于 PAM50 亚型区扩增峰的基因的顺式效应热图。(e) 通过 SNF 使用 mRNA 和蛋白质丰度对肿瘤样本进行集成聚类。(f-g) Kaplan-Meier 曲线显示分配给总体中每个集成聚类的样本的生存结果。 (h-i) 多变量Cox回归分析的森林图,对整体人群和HR HER2 -亚组的综合聚类、肿瘤大小和淋巴结状态进行无复发生存分析。
04
综合代谢组学分析揭示铁死亡是基底样肿瘤的潜在治疗靶点
他们在CBCGA队列中定量了669种极性代谢物和1312种脂质(图4a)。在极性代谢组学和脂质组学的t分布随机邻居嵌入(t-SNE)图中,肿瘤样本和正常样本明显分离,表明肿瘤中存在明显的代谢失调(图4b)。差异代谢物的分析表明,与健康组织相比,大多数代谢物在肿瘤中表现出富集。值得注意的是,氨基酸和多肽在肿瘤中显著上调,表明蛋白质合成旺盛。为了进一步分析极性代谢组学,他们进行了网络分析,以鉴定高度连接的代谢蛋白和极性代谢产物。然后,对这些蛋白质和代谢物的丰度进行注释。高度连接的代谢蛋白和极性代谢物通常也具有亚型特异性,这表明乳腺肿瘤的每个固有亚型都具有不同的极性代谢组学特征(图4c)。他们在管腔型和HER2富集的肿瘤中观察到神经酰胺代谢相关蛋白和代谢物的上调,在HER2富集的肿瘤和基底细胞样肿瘤中观察到色氨酸和烟酰胺腺嘌呤二核苷酸分解代谢的激活,在基底细胞样肿瘤中观察到谷氨酰胺转化为N-乙酰-谷氨酸的富集(图4d)。
在脂质水平方面,他们综合比较了内在亚型的脂质亚类丰度,发现鞘脂在管腔肿瘤中富集,而甘油磷脂在HER2富集和基底样肿瘤中富集,这可能表明这些肿瘤细胞生长分裂旺盛(图4e)。特别是,氧化磷脂在基底细胞样肿瘤中统计富集最多,提示对该亚型铁死亡的潜在脆弱性(图4f-g)。
基底样肿瘤含有大量与铁死亡相关的多不饱和脂肪酸(PUFA)、含PUFA磷脂(PL-PUFA)和氧化磷脂(PL-PUFA-OOH)(图4h)。关键的铁死亡相关蛋白,如ACSL4和TFRC,在基底样肿瘤中也上调。同样,基底细胞样亚型的类器官对铁死亡诱导性物质Erastin和RSL3更敏感(图4i)。综上所述,系统分析评估了乳腺癌中的代谢失调,并揭示了铁死亡可能是基底样肿瘤的潜在治疗靶点。
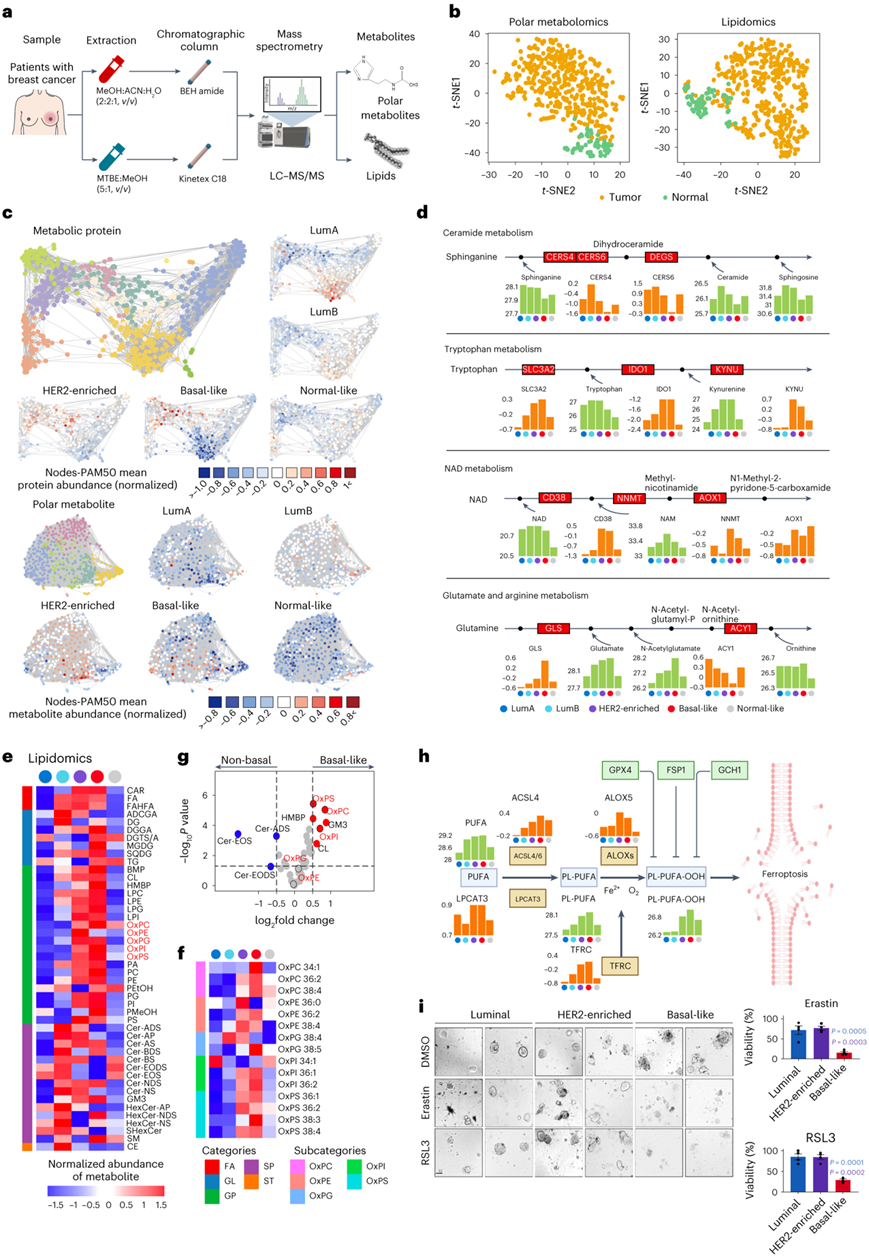
图4. 利用极性代谢组学和脂质组学对代谢失调进行系统评估。
(a) 总结代谢物分析工作流程的示意图。 (b) 极性代谢组学(左)和脂质组学(右)的t -SNE 图。 (c) 基于606个蛋白质的乳腺癌代谢蛋白质相关网络。 (d) c中出现的代表性代谢差异显示在根据KEGG通路图修改的通路模块。 (e) 按脂质亚类分类的亚型特异性脂质的图示。 (f) 按脂质亚类分类的亚型特异性磷脂氢过氧化物的图示。(g) 火山图说明了基底样乳腺癌中亚型特异性脂质亚类。 (h) 示意图总结了铁死亡的关键过程以及PAM50亚型之间与铁死亡相关的关键脂质和蛋白质的比较。 (i) 比较铁死亡诱导剂Erastin和RSL3在不同亚型的患者来源的类器官中的功效。
05
免疫基因组分析揭示了乳腺癌中TME的异质性
利用乳腺癌的转录组学数据,估计了22种不同免疫细胞类型和2种基质细胞类型的丰度水平。基于这些结果,他们确定了乳腺癌中三种不同的肿瘤微环境(TME)表型。“冷”TME显示相对较低的免疫细胞浸润。“中度”TME是通过基质细胞浸润和一些无活性先天免疫细胞的存在来区分的。“热”TME表现出高丰度的适应性和活跃的先天免疫细胞,表明强烈的抗肿瘤免疫反应。与其他两种TME表型相比,“热”TME具有更高的免疫治疗反应预测得分(图5b)。他们还探讨了 PAM50 内在亚型中 TME 表型的分布(图5c)。基底样肿瘤和富含 HER2 的肿瘤具有较高比例的“热”TME 亚型(富含 HER2 的肿瘤为 56.8%,基底样肿瘤为 50.9%)。此外,虽然大部分管腔亚型分为“冷”和“中度”TME 亚型,但 31.5% 的管腔 B 肿瘤归入“热”亚型。
“冷”和“中度”TME 亚型的 T 细胞受体 (TCR) 多样性较低,克隆性较高,这表明通过低 T 细胞扩增实现免疫逃逸机制;然而,B细胞受体(BCR)多样性和克隆性的分布在乳腺癌的不同TME亚型之间存在差异,表明BCR库的功能存在异质性(图5d)。就肿瘤抗原而言,具有“冷”TME的基底样肿瘤比具有“热”TME的基底样肿瘤表现出更高水平的肿瘤抗原产生(图5d);然而,该亚型的免疫浸润较低,促使人们对该亚型的抗原呈递系统进行探索。相应地,“冷”TME 的基底样肿瘤主要组织相容性复合体基因的表达较低,杂合性丢失(LOH)和等位基因失衡的比例较高,这证明了乳腺“冷”肿瘤的组织相容性复合体基因表达略高(图5e)。
此外,他们进行了上游基因组分析,以确定不同TME表型的突变和CNA特征(图5f-g)。TP53(53.8%)、XIPR2(5.3%)和LRP1B(4.9%)在这些肿瘤中最常见,而GATA3(4.5%)在这些肿瘤中最罕见。此外,CSMD1(5.6%)在“冷”肿瘤中最常检测到。对于CNAs, 17q12的扩增/获得(51.0%)和8p22的缺失 (69.5%)在“热”肿瘤中富集,而16p13.3的扩增(45.9%)和16q24.2的缺失 (39.1%)在该表型中最罕见。
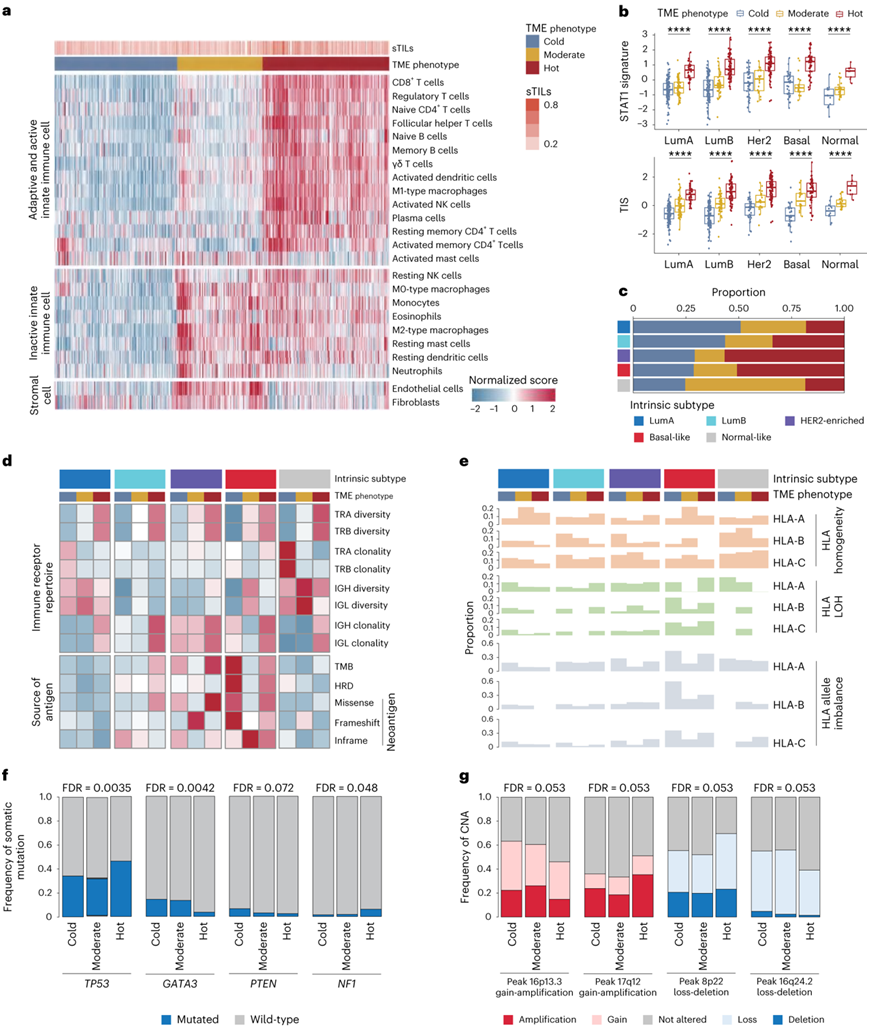
图5. 免疫基因组分析破译了乳腺癌 TME 的异质性。
(a) k均值聚类根据 24 种微环境细胞类型的估计丰度揭示了三种不同的 TME 表型。 (b) 每个指定 PAM50 亚型中 TME 表型的免疫治疗反应预测评分的比较。(c) PAM50 亚型中 TME 表型的分布。 (d-e) 几种免疫基因组学特征的比较,包括免疫受体谱和肿瘤抗原产生和HLA等位基因状态在每个PAM50亚型的TME表型中。(f-g) 体细胞突变和 CNA与 TME 表型之间的关联。
06
使用机器学习方法的多模式数据集成将乳腺癌患者分为不同的复发风险组
复发风险分层和预后预测是乳腺癌临床管理和转化研究的主要问题。他们整合了从微观到介观尺度的数据维度,以全面了解乳腺癌的生物学特征,提高预后预测的效果(图6a)。他们使用分层随机抽样方法从拥有所有维度完整数据的总共 198 人中选择了 80 人的测试队列,其中考虑了复发事件(80 个选定病例中发生了 17 个事件)。然后,他们使用不同的组学组合开发了一系列预后预测模型(图6b)。所有模型测试都是在这个共有的独立测试集中进行的,而剩余的样本在构建每个模型时分配给训练队列。预测模型采用多步骤流程,首先通过共线性约简和单变量选择对特征进行筛选。他们采用了Cox 比例风险模型、随机生存森林模型和生存支持向量机模型,这三种算法同时运行。对三种算法生成的分数进行平均以创建最终的风险分数。通过实施五重交叉验证方法来优化模型超参数。训练后的模型在独立的 80 个样本测试队列中得到验证(图6c)。在测试队列中,多模态模型结合了转录组数据、代谢组数据、病理特征、IHC亚型和临床分期(TMPIC)的模式,C-index为0.78。TMPIC模型在训练集(图6d)和测试集中对复发风险高和低的患者进行了分层(图6e)。TMPIC模型包含5个基因、5个代谢物、5个数字病理特征、IHC亚型和临床肿瘤分期(图6f)。
在这些基本特征中,一些整合聚类特异性基因表达(TFF3和GFRA1)、代谢物(硫鸟嘌呤、苯丙氨酰甲硫氨酸和磷脂酰乙醇胺)和数字病理特征(Percentage_Normal和Texture_IntensityMean_Kurtosis)与良好预后相关,而其他复发风险相关基因(CCNB1、CTSV和CDH3)、代谢物(2-苯基丙酸和磷脂酰甘油)和病理特征(Texture_IntensityMax_Mean 和 Texture_IntensityMax_Skew)与不良结果相关。总之,本项研究建立了一个全面的工作流程,并证明了多模式整合在预测乳腺癌预后方面的潜力。
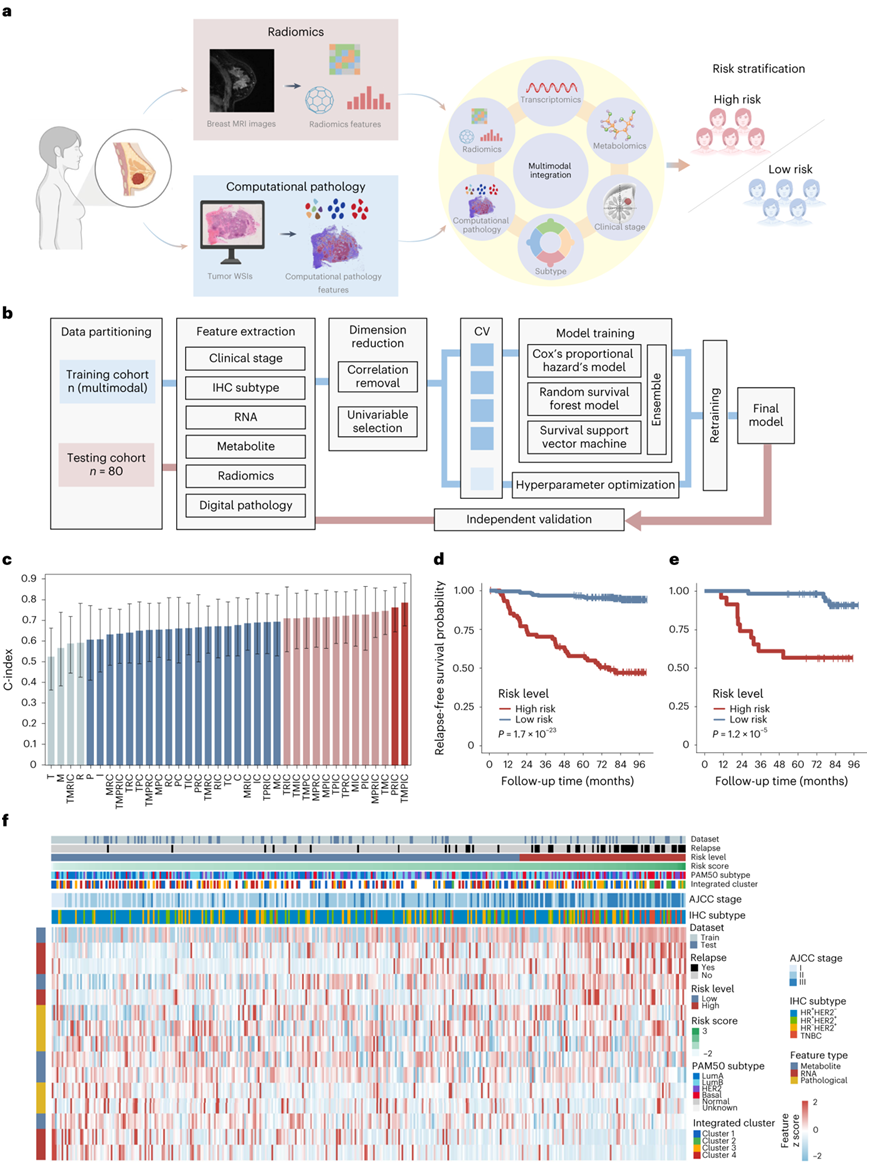
图6. 使用机器学习进行乳腺癌风险分层的多模式数据集成。
(a) 用于风险分层的多模式数据集成示意图。 (b) 用于风险分层的多模式集成模型开发的机器学习框架。 (c) 结合多模态特征的模型的 C 指数,用于对测试队列中的患者预后进行分层。(d-e)在训练组和检验组中比较由TMPIC模型确定的高危组和低危组。 (f) 热图显示了TMPIC模型中使用的多模态特征的z分数。
+ + + + + + + + + + +
结 论
本项研究建立了一个包含434例中国HER2低表达乳腺癌患者(433例女性和1例男性)的队列,并整合了基因组、转录组、蛋白质组和代谢组学数据。在这一队列中,在激素受体阴性亚组中,HER2-low肿瘤与HER2-0肿瘤的区别更明显。在HER2-low肿瘤中,激素受体阴性亚组也存在显著的患者间异质性:基底细胞样型肿瘤类似于HER2-0疾病,而非基底细胞样型HER2-low肿瘤类似于HER2阳性疾病。这些非基底细胞样HER2-low肿瘤富集于HER2富集亚型和雄激素受体管腔亚型,并具有PIK3CA突变、FGFR4/PTK6/ERBB4过表达和脂质代谢激活的特征。在激素受体阳性的肿瘤中,HER2-low肿瘤在17q峰的缺失比HER2-0肿瘤少。这项研究揭示了HER2-low乳腺癌的异质性,并强调需要对激素受体状态和分子亚型进行更精确的分层。
+ + + + +



