

 English
English文献解读|Nature(50.5):疾病相关基因沙漠通过 ETS2 调控巨噬细胞炎症
✦ +
+
论文ID
原名:A disease-associated gene desert directs macrophage inflammation through ETS2
译名:疾病相关基因沙漠通过 ETS2 调控巨噬细胞炎症
期刊:Nature
影响因子:50.5
发表时间:2024.06.05
DOI号:10.1038/s41586-024-07501-1
背 景
自身免疫性和炎症性疾病的发病率不断上升,对人类健康构成了日益严重的威胁。从克罗恩病和溃疡性结肠炎[统称为炎症性肠病 (IBD)]到牛皮癣和狼疮,都需要更好的治疗方法。现有治疗方法的疗效有限以及药物开发过程中的高失败率使这一问题更加严重,迫切需要更好地了解疾病机制。
实验设计
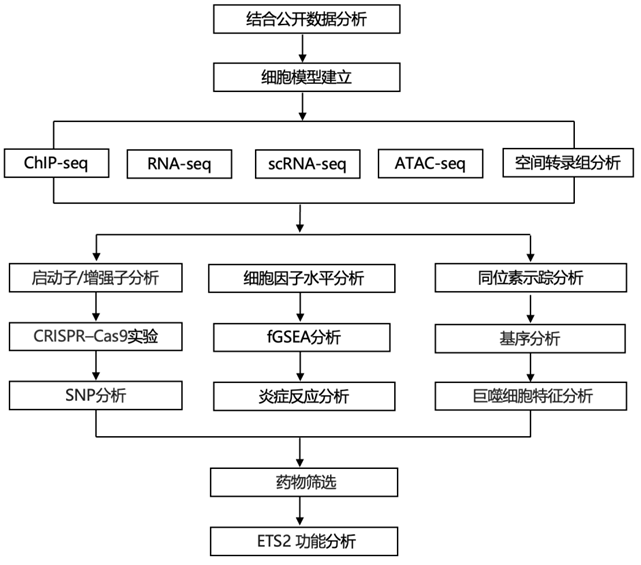
结 果
01

chr21q22 的分子机制分析
一些遗传变异可导致多种疾病,这凸显了它们的生物学重要性以及研究共同疾病机制的机会。值得注意的是,21q22 染色体 (chr21q22) 上的基因间区域,其中主要等位基因单倍型导致五种炎症疾病,此类区域最初因缺乏编码基因而称为“基因沙漠 (gene deserts) ”。 为了分析共同的疾病机制,研究团队进行了共定位分析,并证实每种疾病的遗传基础都是相同的,这意味着由共同的致病变异和共同的分子效应导致(图 1a)。由于这些异质性疾病都是免疫介导的,他们推断该基因座一定包含在免疫细胞中发挥作用的远端增强子。通过检测H3K27ac 染色质免疫沉淀测序 (ChIP-seq) 数据(该数据标记了活性增强子和启动子),他们在该基因座内发现了单核细胞/巨噬细胞特异性增强子(图1b)。单核细胞和巨噬细胞在许多免疫介导疾病中发挥关键作用,产生的细胞因子通常是治疗的靶点。
接下来,他们试图识别由该增强子调控的基因。虽然相关基因座缺乏编码基因,但之前的研究中已经重点介绍了几个附近的候选基因,包括PSMG1 、BRWD1和ETS2(图1a)。利用启动子捕获 Hi-C 和人类单核细胞的表达定量基因座 (eQTL) 数据,他们发现与疾病相关的基因座与ETS2的启动子[最远的候选基因(约290 kb)]发生物理相互作用,并且风险单倍型与较高的ETS2表达相关(图1c)。为了直接证实ETS2是致病因素,他们使用 CRISPR–Cas9 技术敲除了原代人类单核细胞中的 1.85 kb 增强子区域,然后将这些细胞与炎症配体一起培养,包括 TNF(促炎细胞因子)、前列腺素 E2(促炎脂质)和 Pam3CSK4(TLR1/2 激动剂)(TPP 模型)(图1d)。该模型(TPP模型)旨在模拟慢性炎症,并且比经典的 IFNγ 驱动或 IL-4 驱动模型更好反映疾病巨噬细胞状态。由于没有针对候选基因的流式细胞分析抗体,他们使用 PrimeFlow 技术分析了 mRNA 表达的动态,并检测到TPP 刺激未编辑的单核细胞后所有三个基因(ETS2、BRWD1和PSMG1)的水平均增加(图1e)。敲除chr21q22 增强子不会影响BRWD1或PSMG1 的表达,但ETS2的上调显著降低(图1f),证实该多效性位点包含远端ETS2增强子。
为了识别出致病变异,他们在大型 IBD全基因组关联研究 (GWAS)中进行了统计分析。不幸的是,由于候选单核苷酸多态性 (SNP) 之间存在高度连锁不平衡,他们采用大规模并行报告基因检测 (MPRA)方法,首先描绘出基因座上的活性增强子,然后评估是否有任何候选 SNP 可能改变增强子活性(图1g)。使用滑动窗口分析,他们确定了一个 442 bp 增强子活性焦点(染色体 21:40466236–40466677,hg19)(图1h),其中包含三个(七个中的)候选 SNP。其中两种多态性是转录惰性的,但第三种(rs2836882)具有所有候选 SNP 中最强的表达调节作用,风险等位基因(G)增加转录,与ETS2 eQTL 一致(图1h)。该 SNP 位于每个共定位分子性状的可信集中,位于巨噬细胞 PU.1 ChIP-seq 峰内(图1i)。PU.1 是髓细胞中的非经典先驱因子,可与 DNA 结合,启动染色质重塑(从而使其他转录因子结合)并激活转录。为了确定 rs2836882 是否可能影响 PU.1 结合,他们确定了来自杂合巨噬细胞的 PU.1 ChIP-seq 数据并检测了结合中的等位基因不平衡。虽然不位于典型的 PU.1 基序内,但仍检测到了强的等位基因特异性结合,与 rs2836882 风险等位基因的结合率高出四倍多(图1i-j)。此外,对rs2836882杂合子的单核细胞和巨噬细胞进行的转座酶可及染色质测序(ATAC-seq)分析表明,染色质可及性存在等位基因差异,这与先驱因子的差异结合一致(图1k)。为了检测内源性基因座的等位基因特异性增强子活性,他们对来自rs2836882主要和次要等位基因纯合子的炎症巨噬细胞进行了H3K27ac ChIP-seq分析。虽然大多数chr21q22增强子峰在这些供者中相似,但在主要等位基因纯合子中,覆盖rs2836882的增强子活性明显更强,导致整个位点的活性增加了大约2.5倍(图1l)。综上所述,这些数据揭示了一种机制,即通过在原代巨噬细胞中的功能效应确定的chr21q22的假定因果变异体促进先驱因子的结合,增强染色质可及性,并增加远端ETS2增强子的活性。
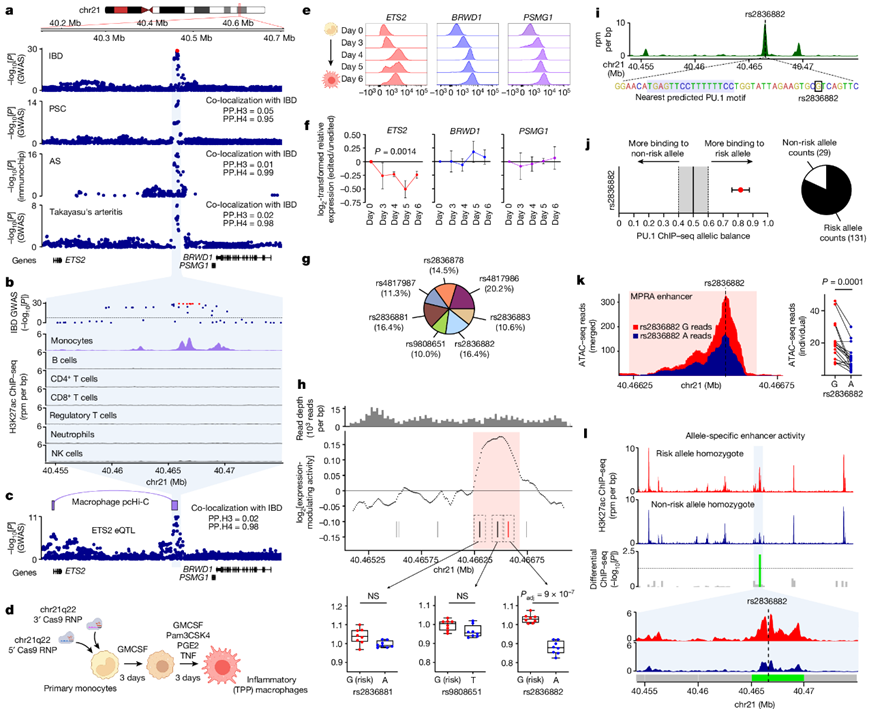
图1. 分析chr21q22 处的分子机制。
(a) chr21q22 处的疾病关联。(b) chr21q22 处的免疫细胞 H3K27ac ChIP-seq。(c) 静息单核细胞中的ETS2 eQTL,与 IBD 关联共定位。(d) 研究炎症 (TPP) 巨噬细胞中 chr21q22 基因座的实验示意图。(e) 使用 PrimeFlow RNA 检测测量 TPP 刺激期间ETS2、BRWD1和PSMG1 mRNA 表达。(f) chr21q22 编辑的巨噬细胞与未编辑细胞中的相对ETS2、BRWD1和PSMG1表达。(g) SuSiE 精细映射 chr21q22 处 IBD 相关 SNP 的后验概率。(h) chr21q22 处的巨噬细胞 MPRA。(i) 炎性巨噬细胞 PU.1 ChIP-seq 在 chr21q22 处达到峰值。(j) 在两个杂合巨噬细胞数据集中对 rs2836882 处等位基因特异性 PU.1 ChIP-seq 结合进行 BaalChIP 分析。(k) 16 位杂合供体(包括健康对照和强直性脊柱炎患者)单核细胞中 rs2836882 处的等位基因特异性 ATAC-seq 读数。(l) rs2836882 处风险(上)或非风险(下)等位基因纯合子的 H3K27ac ChIP-seq 数据。
02
巨噬细胞炎症需要 ETS2
为了阐明ETS2在人类巨噬细胞中的作用,并确定失调的ETS2表达如何导致疾病,他们首先使用基于 CRISPR- Cas9的功能丧失方法(图2a)。相比之下,ETS2敲除后,促炎细胞因子(包括IL-6、IL-8和IL-1β)的产生显著减少(图2b),而IL-10(一种抗炎细胞因子)受影响较小。使用可通过流式细胞检测的荧光标记粒子,发现ETS2破坏后吞噬作用同样受损(图2c)。破坏ETS2可显著降低巨噬细胞的氧化爆发——很可能是通过降低关键 NADPH 氧化酶成分的表达来实现的(图2d)。
为了了解这些影响的分子基础,他们对来自多个供体的 ETS2编辑和未编辑的炎症巨噬细胞进行了转录组分析(RNA-seq)。破坏ETS2会导致广泛的转录变化,许多炎症基因的表达降低(图2e)。这些包括细胞因子(如TNFSF10/TRAIL、TNFSF13、IL1A和IL1B)、趋化因子(如CXCL3、CXCL5、CCL2和CCL5)、分泌的效应分子(如S100A8、S100A9、MMP14和MMP9)、细胞表面受体(如 FCGR2A、FCGR2C和TREM1)、模式识别受体 (如TLR2、TLR6和NOD2)和信号分子(如MAP2K、GPR84和NLRP3)。为了更好地表征受影响的通路,他们使用基因本体 (GO) 生物通路数据集进行了基因集富集分析 (fGSEA),这证实了功能缺陷,其中负富集最多的通路(因ETS2破坏而下调)与巨噬细胞活化、炎性细胞因子产生、吞噬作用和 ROS 产生有关(图2f)。ETS2干扰后上调的基因较少(图2e),但观察到有氧呼吸和氧化磷酸化(OXPHOS)—与抗炎表型相关的代谢过程富集(图2f)。如预期的那样,chr21q22 增强子的缺失表型复制了破坏ETS2的转录和功能效应(图2g)。
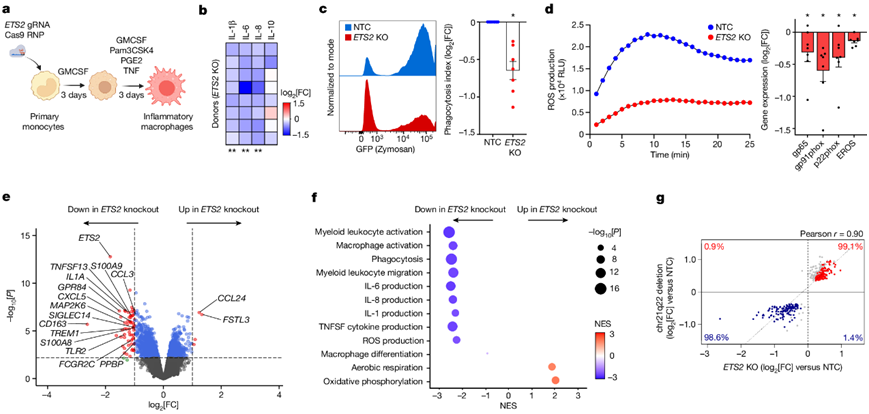
图2. ETS2对巨噬细胞炎症反应至关重要。
(a) 研究炎性(TPP)巨噬细胞中ETS2 的实验示意图。(b) ETS2 破坏后的细胞因子分泌。ETS2编辑与未编辑巨噬细胞的相对细胞因子水平热图。(c) 对荧光标记的酵母聚糖颗粒的吞噬作用以及吞噬指数。(d) ETS2编辑和未编辑巨噬细胞产生的 ROS 。(e) ETS2 编辑与未编辑的 TPP 巨噬细胞中差异表达基因的 RNA-seq 分析。(f) ETS2编辑和未编辑的 TPP 巨噬细胞之间差异表达基因的 fGSEA。(g) chr21q22 增强子缺失差异表达基因。
03
ETS2 协调巨噬细胞炎症
他们接下来研究了增加ETS2表达的影响,因为这是驱动疾病风险的因素。为此,他们优化了一种在原代巨噬细胞中控制靶基因过表达的方法,通过转染体外转录的 mRNA,将修饰的 mRNA 最大程度地降低免疫原性(图3a)。静息的、未活化的巨噬细胞经转染ETS2 mRNA 或其反向补体,从而控制 mRNA 的数量、长度和嘌呤/嘧啶组成 (图3b)。转染后,细胞暴露于低剂量脂多糖可能会加剧炎症反应(图3a)。他们发现过表达ETS2会增加促炎细胞因子的分泌,而 IL-10 受到的影响较小。为了更好地描述这种反应,他们进行了RNA-seq并重新检测了涉及ETS2 的炎症通路。值得注意的是,所有这些通路(包括巨噬细胞活化、细胞因子产生、ROS 产生、吞噬作用和迁移)都是通过ETS2过表达以剂量依赖性方式诱导的,当转染更多的ETS2 mRNA时,每种通路的富集程度都会更高(图3c)。这表明ETS2对于人类巨噬细胞的炎症反应是至关重要,与作为效应功能的中心调节因子一致,其失调与疾病直接相关。
为了测试ETS2是否导致疾病中的巨噬细胞表型,他们进行了单细胞转录组分析(scRNA-seq),比较了静息巨噬细胞中ETS2过表达的影响与克罗恩病中肠道巨噬细胞的特征。ETS2过表达诱导的转录状态与疾病巨噬细胞非常相似,大多数特征基因的富集,包括几个治疗靶点(图3d)。在其他 chr21q22 相关疾病的髓系特征中也观察到了类似的富集,在活动性细菌感染中也观察到了类似的富集,但程度较小,但在流感和肿瘤巨噬细胞的特征中没有观察到,这表明 ETS2 并非简单地诱导一般激活(图3e)。值得注意的是,与许多IBD通路相比,IBD相关SNP在巨噬细胞ETS2通路中更显著地富集(图3f)。
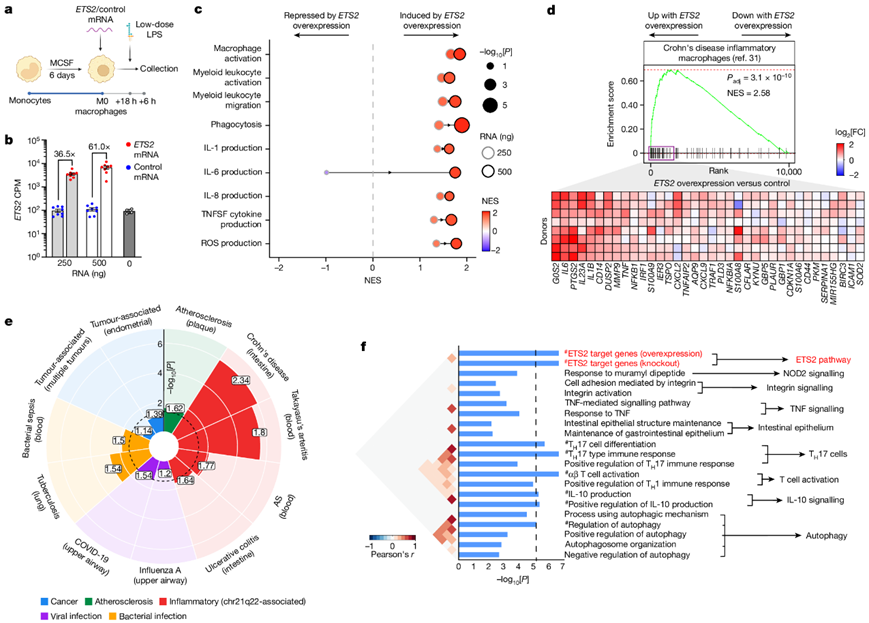
图3. ETS2 协调巨噬细胞炎症反应。
(a) 研究ETS2过表达影响的实验示意图。(b) 巨噬细胞中的ETS2 mRNA 水平。(c) ETS2过表达和对照巨噬细胞之间差异表达基因的 fGSEA 分析。(d) ETS2过表达巨噬细胞(与对照相比)中克罗恩病肠道巨噬细胞特征的 fGSEA 分析。(e) ETS2过表达巨噬细胞中所示疾病患者的巨噬细胞特征富集。(f) ETS2调控基因(红色)和已知 IBD 通路(黑色)内 241 个 IBD SNP 标记基因的 SNPsea 分析。
04
ETS2 具有明显的炎症作用
他们鉴定了在 67 种人类巨噬细胞活化条件下(包括 28 种刺激和不同的暴露持续时间)与ETS2共表达的基因,其中PFKFB3(编码糖酵解的限速酶)为最高度共表达的基因,HIF1A也高度共表达(图4a),这些基因共同促进了髓系炎症反应所必需的“糖酵解开关”。 因此,他们假设ETS2可能通过代谢重编程控制炎症——OXPHOS 基因与ETS2呈负相关(图4a)。
为了评估破坏ETS2的代谢后果,他们使用气相色谱-质谱法 (GC-MS) 定量了编辑和未编辑的 TPP 巨噬细胞中13C-葡萄糖的标记掺入量。ETS2破坏后检测到标记和总葡萄糖代谢物普遍适度减少(图4b),这导致糖酵解和三羧酸 (TCA) 循环代谢物,乳酸(无氧糖酵解的标志)和琥珀酸(关键的炎症代谢物)显著减少。这些结果与糖酵解抑制一致,TCA 代谢物的减少是由于进入 TCA 的流量减少和线粒体 OXPHOS 的消耗增加。
为了确定代谢变化是否是 ETS2 介导的炎症效应的原因,他们用罗沙司他(一种促进糖酵解的 HIF1α 稳定剂)处理了ETS2编辑的巨噬细胞。这对糖酵解和 OXPHOS 基因产生了预期的影响,但并没有从转录或功能上挽救ETS2破坏的影响(图4c)。因此,他们重新考虑是否可以直接识别 ETS2 靶基因。由于 ChIP-seq 涉及可改变蛋白质表位并阻止抗体结合的步骤(例如固定),他们检测了是否有任何抗 ETS2 抗体可以用于靶标下的切割并使用核酸酶释放(CUT&RUN),鉴定出多个显著富集的基因组区域(峰),其中 6560 个在两个生物学重复中可重复检测到(图4d)。这些峰大多位于活性调控区域(90% 在启动子或增强子中)(图4d-e),并且高度富集典型的 ETS2 基序和已知 ETS2 相互作用因子的基序,包括 FOS、JUN 和 NF-κB(图4f)。结合生物学重复以改进峰值检测后,他们发现 ETS2 与参与多种炎症功能的基因结合,包括NCF4(ROS 生成)、NLRP3(炎症小体活化)和TLR4(细菌模式识别)(图4g)。总体而言, ETS2破坏后失调的基因中有 48.3%和ETS2过表达后失调的基因中有 50.3%,在其核心启动子或顺式调控元件内包含 ETS2 结合峰(图4h)。值得注意的是,ETS2 靶标包括HIF1A、PFKFB3和其他糖酵解基因(如GPI、HK2和HK3),这与观察到的作为该复杂炎症程序的一部分直接诱导的代谢变化一致,他们还检测到 ETS2 与 chr21q22 增强子结合(图4i),表明ETS2 可能有助于其自身增强子的活性。
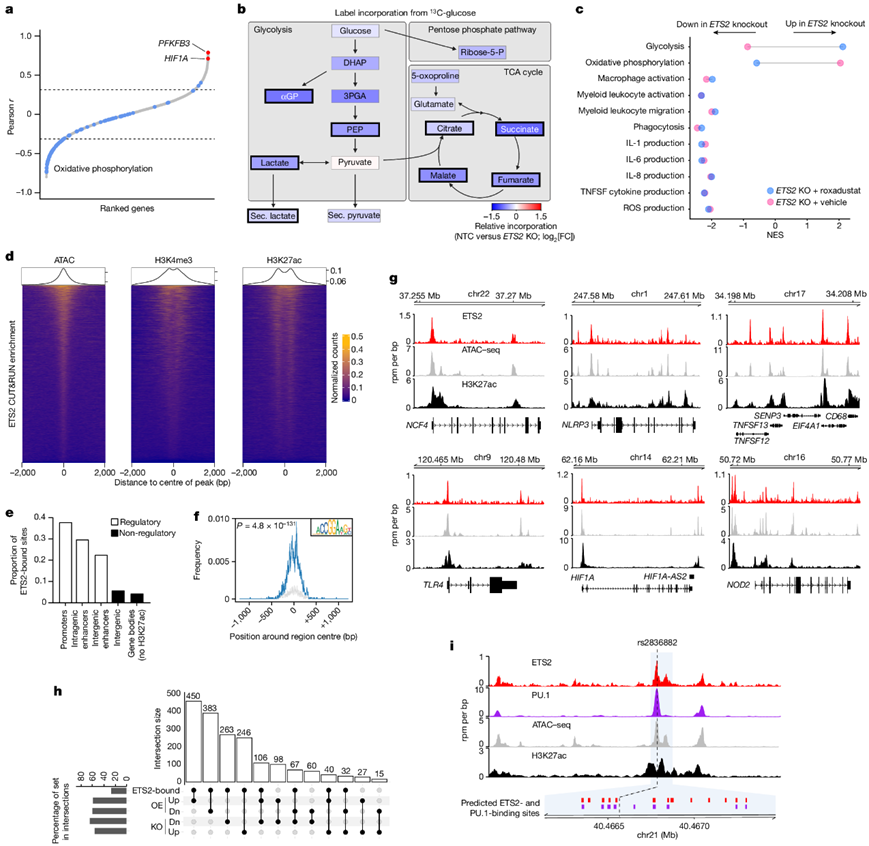
图4. ETS2 通过转录和代谢效应指导巨噬细胞反应。
(a) 在 67 种单核细胞/巨噬细胞活化条件下与ETS2共表达的基因。(b) ETS2破坏对葡萄糖代谢的影响。(c) 差异表达基因的fGSEA分析。(d) 巨噬细胞ETS2 CUT&RUN 峰的富集热图。(e) ETS2 结合位点的功能注释。(f) CUT&RUN 峰中的 ETS2 基序富集。(g) 选定基因座处的 ETS2 结合、染色质可接近性(ATAC-seq)和调节活性(H3K27ac)。(h) 核心启动子或顺式调控元件中 ETS2 峰的基因与ETS2编辑(KO)或过表达(OE)后上调(Up)或下调(Dn)的基因之间的交集。(i) chr21q22 处的 ETS2 结合、PU.1 结合、染色质可及性和增强子活性。
05
针对疾病中的 ETS2 通路
为了评估 ETS2 如何影响患病组织中的巨噬细胞异质性,以及是否可以将其作为治疗靶点,他们检测了克罗恩病患者和健康对照个体的肠道 scRNA-seq 数据。在髓系细胞中,利用已建立的标记物和/或先前的文献检测并鉴定了七个聚类(图5a-b)。炎性巨噬细胞(聚类1,表达 CD209、CCL4、IL1B 和 FCGR3A)和炎性单核细胞(聚类 2,表达 S100A8/A9、TREM1、CD14 和 MMP9)在疾病中扩增,并且表达ETS2和 ETS2 调节基因高于其他聚类,包括组织驻留巨噬细胞(聚类0,表达 C1QA、C1QB、FTL 和 CD63)和常规树突状细胞(聚类5,表达 CLEC9A、CADM1 和 XCR1)(图5a-b)。空间转录组学分析显示,在 PSC 肝组织中观察到炎性巨噬细胞的类似增加,这些细胞与胆管细胞紧密相邻(图5c-e)。值得注意的是,巨噬细胞越靠近胆管细胞,ETS2 调控基因的表达就越高(图5f)。
他们使用了 NIH LINCS 数据库来识别可能调节 ETS2 活性的药物。它包含来自暴露于约 6000 种小分子的细胞系的 30000 多个差异表达基因列表。使用 fGSEA,906 个特征模拟了破坏ETS2的效果。最大的药物类别是 MEK 抑制剂(图5g),它们受批准用于治疗非炎症性人类疾病(如神经纤维瘤病)。这个结果不是由于单一化合物,而是由于多种 MEK1/2 抑制剂下调 ETS2 靶基因的类效应(图5h),这在生物学上是有意义的,因为 MEK1/2 和已鉴定的其他几个靶点都是已知的 ETS 家族转录因子的调节剂(图5g)。为了检测MEK 抑制是否能消除人类巨噬细胞中 ETS2 驱动的炎症,他们用选择性非 ATP 竞争性 MEK 抑制剂 PD-0325901 处理 TPP 巨噬细胞,观察到强效的抗炎活性,其表型模拟了破坏ETS2或 chr21q22 增强子的效果(图5i-j)。MEK 抑制可将炎症细胞因子释放降低至与英夫利昔单抗(一种广泛用于治疗 IBD 的抗 TNF 抗体)类似的水平(图5k)。此外,ETS2 调节的基因表达降低(图5l),转录炎症评分改善(图5m)。总之,这些数据表明,针对ETS2上游调节剂可以消除 chr21q22 相关疾病中的病理性炎症,并且可能具有治疗作用。
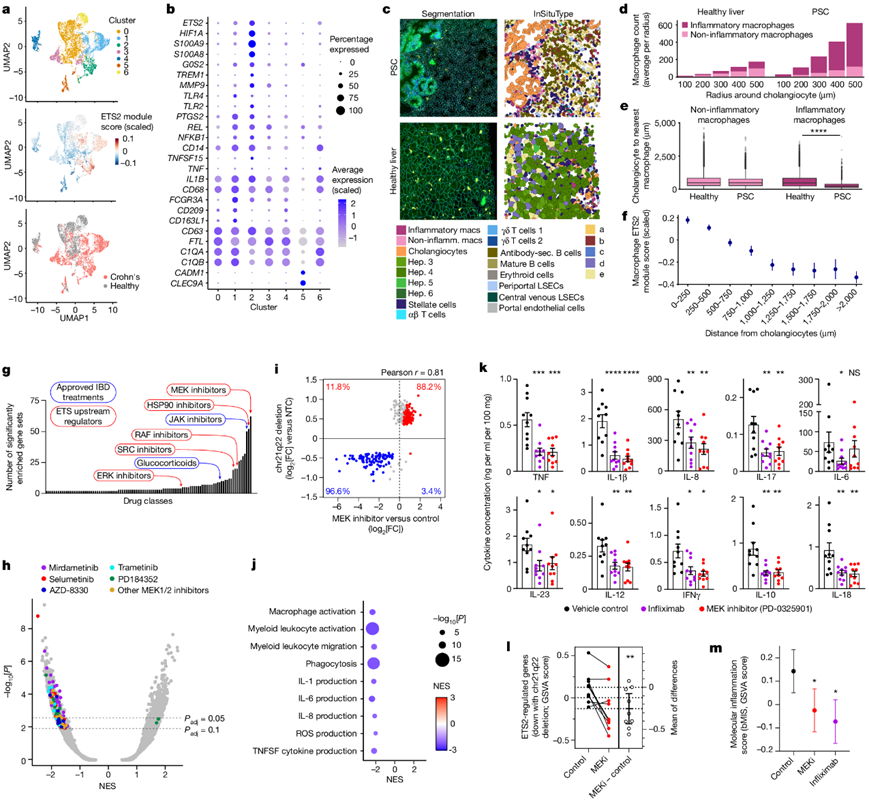
图5. ETS2 驱动的炎症在疾病中很明显,可以作为治疗的针对性指标。
(a) UMAP可视化。 (b) 选定基因的表达分析。(c) PSC 和健康肝脏的空间转录组学。(d) 胆管细胞指示距离内的巨噬细胞数量。(e) 从胆管细胞到最近的巨噬细胞的距离。(f) 在距离胆管细胞确定的距离处的 21067 个 PSC 巨噬细胞中 ETS2 调节基因的表达。(g) 表型模拟ETS2破坏的药物类别。(h) fGSEA 的 NIH LINCS 药物特征结果。(i) chr21q22 增强子敲除后差异表达基因。(j) MEK 抑制剂处理和对照 TPP 巨噬细胞之间差异表达基因的 fGSEA。(k) 使用 PD-0325901、英夫利昔单抗或载体对照的 IBD 活检细胞因子释放。(l)MEK 抑制后 IBD 活检中 chr21q22 下调基因的 GSVA 富集分数。(m) 活检衍生的分子炎症评分(bMIS)的 GSVA 富集分数。
+ + + + + + + + + + +
结 论
本项研究发现致病基因ETS2是人类炎症巨噬细胞的中心调节因子,并探究了扩增ETS2表达的共同疾病机制。ETS2 调控的基因在患病组织中显著表达。在静息巨噬细胞中过度表达ETS2可重现 chr21q22 相关疾病中观察到的炎症状态,并上调多个药物靶点,包括 TNF 和 IL-23。利用细胞特征数据库,确定了可能调节该通路的药物,并在体外和离体验证了一类小分子的强效抗炎活性。总之,这说明了功能基因组学的强大功能,它直接应用于原代人类细胞,可以识别免疫介导的疾病机制和潜在的治疗机会。
+ + + + +



