

 English
English文献解读|Science(44.7):确定驱动 KRAS 突变癌症的 ERK 调节磷酸化蛋白质组
✦ +
+
论文ID
原名:Determining the ERK-regulated phosphoproteome driving KRAS-mutant cancer
译名:确定驱动 KRAS 突变癌症的 ERK 调节磷酸化蛋白质组
期刊:Science
影响因子:44.7
发表时间:2024.06.07
DOI号:10.1126/science.adk0850
背 景
KRAS致癌基因的突变激活是胰腺导管腺癌 (PDAC) 生长的主要遗传驱动因素。研究最深入的直接 KRAS 效应物是 RAF 丝氨酸-苏氨酸激酶(ARAF、BRAF 和 RAF1 或 CRAF)。活性 RAS-GTP 结合并促进 RAF 激酶活性的激活。随后,激活的 RAF 磷酸化并激活高度相关的双特异性丝裂原活化蛋白激酶 (MAPK) 激酶 MEK1 和 MEK2 (MEK1/2),而后者又磷酸化并激活高度相关的 ERK1 和 ERK2 (ERK1/2) 丝氨酸-苏氨酸激酶。RAF 和 MEK 的底物利用率受到严格限制,而 ERK 的底物利用率则广泛且多样。ERK 如何驱动 KRAS 突变型癌症生长仍不清楚。
实验设计

结 果
01

ERK1 和 ERK2 支持 KRAS 依赖性生长
激活的 MEK1 而非 AKT1 足以使 PDAC 细胞对突变型 KRAS 的直接药物抑制不敏感。与 MEK1 不同,ERK 的激活突变在癌症中未发现,可能是因为单点突变不足以完全激活,因此充当相对较弱的致癌基因。为了确定 ERK 激活足以支持 KRAS 驱动的 PDAC 生长的程度,研究者团队通过结合先前描述的点突变生成了 ERK1 和 ERK2 的组成性激活变体(83% 同一性),每个点突变单独都会增加 ERK1 和 ERK2 活性(图1A)。在 KRASG12D突变型 PDAC 细胞系 Pa16C 中,每个 ERK1 突变体在强力霉素 (Dox) 诱导下的瞬时表达表明,双突变体 ERK1 R84S/S170D(称为 ERK1 SD)最强烈地诱导 ERK 底物 FRA1 (pFRA1) 的磷酸化(图 S1B)。他们选择了该突变体和 ERK2 的类似突变体(ERK2 R67S/S153D、 ERK2 SD)进行进一步分析(图 1A)。
他们在 KRAS G12C(MIA PaCa-2 和 UM53)或 KRAS G12D (Pa16C)突变型 PDAC 细胞系中建立了 Dox 诱导的 MYC 表位标记的 ERK1 SD或 ERK2 SD表达。Dox 处理刺激 ERK1/2SD表达的水平与内源性 ERK 相当,但不会显著改变 ERK 底物核糖体蛋白 S6 激酶 (pRSK) 的磷酸化或 MYC 的表达,后者由直接 ERK 磷酸化稳定(图 1B)。然后,他们评估了 ERK1SD或 ERK2 SD突变体的表达是否会影响针对 KRAS-ERK MAPK 级联不同节点的药物抑制剂抑制 PDAC 生长的能力。ERK1/2 SD表达细胞对 MEK1/2 选择性抑制剂曲美替尼 (MEKi) 具有抗性,但对 ERK1/2 选择性抑制剂 SCH772984 (ERKi) 保持敏感性 (图1C)。活化的 ERK1/2 SD表达细胞在 MEK 抑制后保持 ERK 依赖性生长。用 KRAS G12C选择性抑制剂(G12Ci)(MRTX1257、adagrasib以及MRTX849 的类似物)或临床候选药物 KRAS G12D选择性抑制剂(G12Di)(MRTX1133) 处理降低了对照细胞中的 MYC 和 pRSK 水平,而 ERK1/2 SD表达可以挽救这一水平(图 1B)。他们观察到仅激活ERK就能导致完全或接近完全的抗生长抑制(图1C)。
为了确定 ERK1 和 ERK2 是否调节不同的信号活动,他们对六种 KRAS G12C/D突变型 PDAC 细胞系中 147 种已确定的信号蛋白和磷酸化事件进行了反相蛋白质阵列 (RPPA) 分析。抑制 ERK1 或 ERK2 后观察到的信号变化的 Spearman 相关性很高,表明信号变化几乎相同,支持以下结论:它们的功能是冗余的而不是不同的(图 1D)。为进一步支持 ERK1 和 ERK2 的冗余功能,他们发现 ERK1 SD或 ERK2 SD的异位表达减弱了几乎所有 G12Di 诱导的转录变化(分别减弱了 92% 和 83%)(图 1E-F)。 siRNA 急性抑制KRAS和ERK引起的信号变化相似,主要区别在于相对强度。总之,这些结果证明ERK1 和 ERK2 在支持 KRAS 依赖性生长方面存在冗余信号输出。
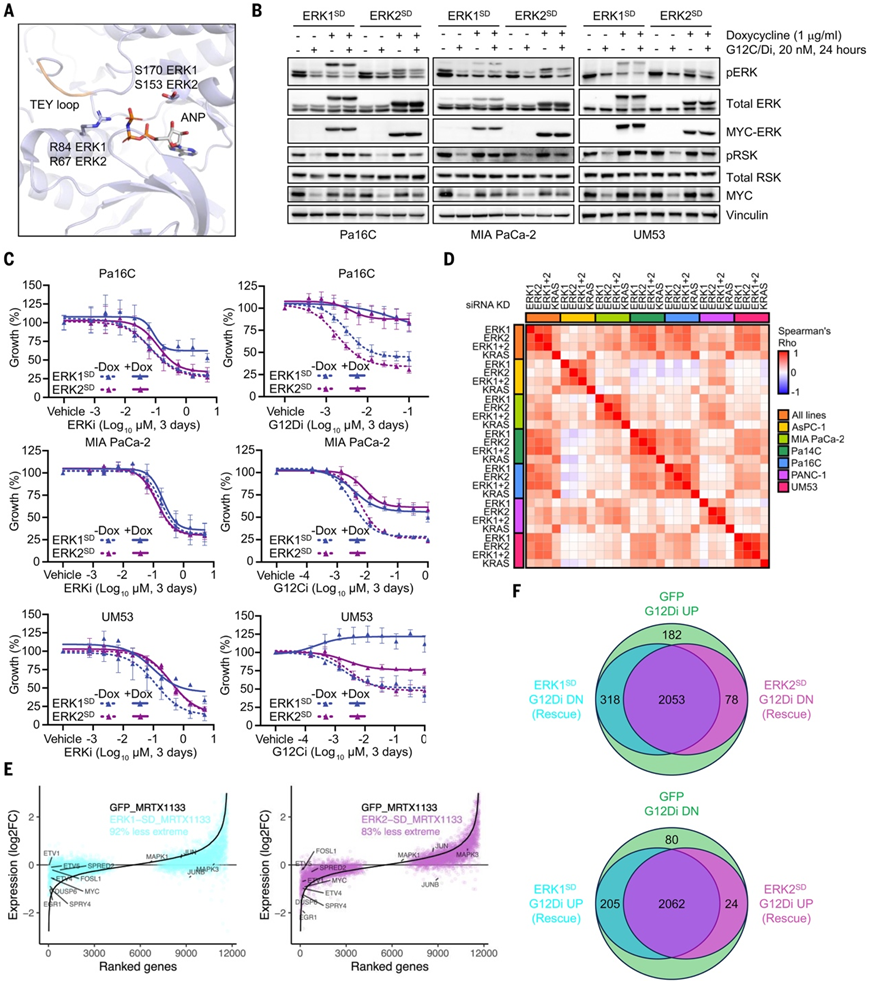
图1. ERK 信号对于 KRAS 调节的 PDAC 生长至关重要。、】(A) ERK2 与 ATP 类似物磷酸氨基膦酸-腺苷酸酯 (ANP) 结合的晶体结构。 (B)免疫印迹分析。(C) 细胞活力测定。(D) RPPA Spearman 相关系数分析。(E) RNA 转录表达变化。(F) 维恩图表示 GFP 表达细胞中 KRAS G12D抑制上调 (G12Di UP) 或下调 (G12Di DN) 的基因。
02
KRAS 突变型 PDAC 中的 ERK 依赖性磷酸化蛋白质组
他们使用了三磷酸腺苷 (ATP) 竞争性和变构性 ERKi SCH772984,体外研究表明,纯化的重组蛋白激酶对 ERK1/2 具有高度选择性。为了评估其在 PDAC 细胞培养中的选择性,他们使用了成熟的多重激酶抑制剂珠和质谱 (MIB-MS)进行检测,评估了一组用 SCH772984 急性(1 小时)或长期(24 小时)处理的六种 KRAS G12C/D突变 PDAC 细胞系(图 2A)。使用 MIB-MS,他们发现在两个时间点量化的 207 种蛋白激酶中,只有 ERK1 和 ERK2 受到选择性抑制(图 2B)。因此,证实了 SCH772984 对 ERK1 和 ERK2 的细胞选择性。为了识别 KRAS 突变型人类癌症中 ERK 依赖性磷酸化蛋白质组,他们使用磷酸肽富集和液相色谱-串联质谱 (LC-MS/MS) 在同一组六种KRAS突变型 PDAC 细胞系中进行了定量磷酸化蛋白质组学研究(图 2C)。为了优化直接 ERK 底物的检测,他们对细胞进行了 1 小时的急性处理,然后进行了长期 24 小时 ERKi 处理,这是由于 ERK 负反馈调节丧失而开始出现补偿活动的时间点,在所有六种细胞系中检测到了总共 13646 个不同的总磷酸位点。其中,932 个在 ERKi 1 小时时受到差异调节,4288 个在 24 小时后受到差异调节(图 2D)。在 1 小时时,磷酸位点主要发生下调,而到 24 小时后,相当一部分发生上调。
将他们的 PDAC ERK 依赖性磷酸化蛋白质组与 ERK 底物概要进行比较,结果显示仅有 12% (580) 个磷酸位点和 33% (707) 个磷酸化蛋白重叠(图 2E)。之后,他们将 ERK 依赖性磷酸化蛋白质组与最近对胰腺癌患者的蛋白质组学研究进行了比较,该研究分析了由 NCI 临床蛋白质组肿瘤分析联盟 (CPTAC) 收集的大部分肿瘤组织(包括相邻的非肿瘤区域)中的蛋白质组和磷酸化蛋白质组。ERK 依赖性磷酸化蛋白质组在磷酸位点和蛋白质水平上与该体内 PDAC 特异性数据集具有更高的重叠程度:分别为 32% 和 77%(图 2E)。不完全重叠可能反映了 PDAC 的细胞含量低,通常仅由 10% 到 15% 的肿瘤组织组成。此外,他们的分析仅包括未经治疗的 PDAC,其中很少是转移性的(26),而他们评估了来自原发性和转移性 PDAC 肿瘤的细胞系。这表明本项研究的数据集更能代表人类 PDAC,并突出了其包含现有数据库中未发现的 2500 多个(>50%)磷酸位点和 200 多个(>10%)蛋白质。
他们还纳入了Kras突变型小鼠 PDAC(KPC;Kras G12D;Trp53 R172H)肿瘤来源的细胞系,以建立跨物种验证,确定了用 KRAS G12C药物抑制剂 (MRTX1257) 处理的 KRAS G12C突变型 H358 非小细胞肺癌 (NSCLC)、SW837 结直肠癌 (CRC) 和 MIA PaCa-2 PDAC 细胞系或用 RAS(ON) 多选择性三复合物抑制剂 RMC-7977 (RASi) 处理的 KPC 小鼠 PDAC 肿瘤来源细胞系的磷酸化蛋白质组。ERK 依赖性磷酸化蛋白质组在人类细胞系中的 KRAS G12C依赖性磷酸化蛋白质组中富集,特别是在下调的磷酸位点中,它占差异调节磷酸位点的 44% 以上(图 2F)。

图2. KRAS突变型PDAC中ERK调控的磷酸化蛋白质组的磷酸化蛋白质组学分析。
(A) 使用 BioRender.com 创建 MIB-MS 实验流程图。(B) 使用 ERKi(SCH772984,1 μM)处理 1 或 24 小时的六种 KRAS 突变细胞系的 MIB-MS 分析。(C) 使用 BioRender.com 创建的蛋白质组学分析实验工作流程。(D) ERK 抑制 1 和 24 小时后磷酸位点的表达差异。(E) PDAC 中 ERK 依赖性磷酸位点或磷酸蛋白与 CPTAC PDAC 数据集、ERK 概要、PSP 调控位点和 PTMsigDB 中报告的磷酸位点或磷酸蛋白之间的重叠。 (F) 差异表达的磷酸位点。
03
ERK 调节富含 ERK 相互作用基序的整体磷酸化蛋白质组
细胞外刺激会促进瞬时 ERK 磷酸化和活化,导致 ERK 从细胞质易位至细胞核。为了评估在突变型 KRAS 持续激活条件下活化 ERK 的亚细胞分布,他们采用了两种方法。首先,使用 ERK1/2 磷酸化特异性抗体进行免疫荧光,该抗体可识别 ERK1/2 TEY 基序处 MEK1/2 的磷酸化。他们检测到整个细胞中的稳态磷酸化 ERK (pERK) 和总 ERK,包括细胞核内的定位(图3A-B)。经过 24 小时的 ERKi 处理后,细胞核内外的pERK 水平都受到强烈抑制。同时,与 ERK 信号的丧失一致,他们观察到 ERK 底物 MYC 表达降低。其次,由于 pERK 可能无法准确监测 ERK 活性,他们使用了一组针对细胞质 (cyto-EKAR4) 或细胞核 (nuc-EKAR4) 区室的 ERK 激酶活性报告基因 (EKAR4)。这些报告基因同样在 24 小时 ERKi 处理后显示出对细胞质和细胞核 ERK 活性的强烈抑制(图3C-D)。
接下来,他们评估了 ERK 依赖性磷酸化蛋白质组,以了解 ERK 调节的磷酸化蛋白质的亚细胞分布,观察到与每个主要细胞器相关的磷酸化蛋白质的变化(图 3E),这与 ERK 调节涵盖所有亚细胞区室的全局磷酸化蛋白质组相一致。在 ERK 抑制 1 小时和 24 小时时,下调的磷酸化蛋白质在细胞骨架蛋白中富集。在 24 小时后,下调的磷酸化蛋白质也在核蛋白中富集,而上调的磷酸化蛋白质则在质膜相关蛋白中富集。
ERK 通过直接的底物相互作用来调节不同的信号通路,这种相互作用会传递到次级的间接相互作用,从而促进亚细胞水平的 ERK 活性。一种可以提供直接ERK底物的近似指示的方法是表征磷酸化蛋白是否存在最小ERK磷酸化基序[S/T]-P,以及两个不同的ERK对接序列,DEF(也称为ERK FXF或F的对接位点)和D(也称为DEJL)基序。这些 ERK 对接位点增强了 ERK 底物结合的特异性和亲和力。在下调的磷酸位点中,在每个时间点所有三个 ERK 定向基序都有富集,其中 ~75% 具有 [S/T]-P 基序,~40% 具有 D 和/或 DEF 基序以及 [S/T]-P(图 3F)。ERK 的亚细胞定位可以指导 ERK 调节的过程,并且 ERK 相互作用基序将 ERK 活性引导至不同亚细胞区室中的底物。因此,他们确定了 ERK 调节的磷蛋白中 ERK 相互作用基序的亚细胞富集情况,并发现了不同的模式,其中 D 基序在每个区室中富集,而 DEF 基序主要定位于核蛋白(图 3G)。然后,他们评估了已确定的 ERK 调节的亚细胞通路中的 ERK 结合和磷酸化基序,发现含有 DEF、D 和 [S/T]-P 基序的蛋白质频率很高。ERKi 处理 1 小时后,在最富集的通路中,43% (16/37) 的细胞骨架相关磷蛋白中发现了 ERK 结合基序,62% (23/37) 的细胞骨架相关磷蛋白含有 [S/T]-P ERK 磷酸化基序(图 3H)。ERKi 处理 24 小时后,在细胞骨架相关磷蛋白中,具有 ERK 结合和 [S/T]-P ERK 磷酸化基序的磷蛋白分别增加到 57% (24/42) 和 81% (34/42)。在 ERKi 处理 24 小时后富集的核相关磷蛋白中,55% (113/204) 含有 ERK 结合基序,72% (286/398) 含有 [S/T]-P 基序。这些结果强调 ERK 可以与整个亚细胞区室中的多种磷蛋白候选物结合,从而影响多种细胞过程。
接下来,他们评估了 ERK 与含 D 和 DEF 基序的蛋白质相互作用在支持 KRAS 依赖性 PDAC 生长中的重要性。点突变选择性地削弱大鼠 ERK2 与含 D 或 DEF 基序的底物相互作用但不削弱其激酶功能。他们将类似的氨基酸取代引入活化的人类 ERK1 SD或 ERK2 SD中,以产生与含 D 基序 [D338N (ERK1 SDN ) 和 D321N (ERK2 SDN )] 或含 DEF 基序 [Y280A (ERK1 SDA ) 和 Y263A (ERK2 SDA )] 底物相互作用受损的变体。然后用编码每个 ERK1/2 突变体的慢病毒载体稳定感染的 Pa16C 细胞用 Dox 处理,以在用各种抑制剂处理之前瞬时表达每个 ERK 突变体(图 3I)。ERK1/2 SD表达细胞对 G12Di 或 MEKi 处理有抗性,但对 ERKi 仍然敏感,而这种抗性在 ERK1/2 SDN/SDA突变体中消失(图 3J)。在用 G12Di 处理的细胞中,ERK1/2 SDN或 ERK1/2 SDA的异位表达无法挽救 ERK 激活(pRSK 和 MYC)(图 3I)。因此,ERK 与 D 和 DEF 基序的相互作用是介导对 KRAS 依赖性生长至关重要的 ERK 信号传导所必需的。

图3. ERK 磷酸化蛋白质组调节由富含 ERK 结合基序的蛋白质组成的多种功能通路。
(A) 免疫荧光的代表性图像。(B) 虚线表示中位数,统计数据表示对至少两次重复的 5 到 10 个视野中的 113 到 655 个单个细胞进行的 Mann-Whitney 检验。(C-D) 用 ERKi (SCH772984, 1 μM) 或 DMSO 处理 4 或 24 小时后,稳定表达非靶向 EKAR4 (EKAR4) 或用核定位信号 (nuc-EKAR4) 或核输出信号 (cyto-EKAR4) 标记的 EKAR4 的 Pa16C 细胞的代表性伪彩色 FRET 强度。(E) ERKi 处理 1 小时和 24 小时后,磷蛋白与差异调节的磷酸位点的亚细胞结合及富集。(F) 磷蛋白中含有 p[S/T]-P 磷酸基序以及 ERK 结合 D 或 DEF 基序的磷酸位点的比例。(G) 磷蛋白与 ERK 结合和磷酸化基序的亚细胞结合。(H) 在 1 小时 ERKi 处理后磷酸位点下调的细胞骨架相关磷蛋白在顶部富集基因集中的磷蛋白成员。(I) 免疫印迹分析。(J) 用剂量递增的 G12Di (MRTX1133) 处理的Dox 诱导的 ERK1 SD、ERK1 SDN和 ERK1 SDA细胞系的活力。
04
ERK 调节复杂的蛋白激酶网络
ERK 底物包括调节基因转录、蛋白质磷酸化和蛋白质稳态的蛋白质,它们驱动 ERK 依赖性二级信号传导,从而改变整体转录组、蛋白质组和磷酸化蛋白质组。因此,他们接下来评估了 ERK 依赖性磷酸化蛋白质组,以了解这些大类蛋白质上的磷酸位点变化(图 4A)。ERK 抑制 1 小时后,发现 33 种表观遗传调节剂、31 种转录因子、28 种激酶、16 种 E3 连接酶和 5 种磷酸酶上的磷酸位点减少。24 小时后,减少的磷酸位点分别增加到 107、98、55、44 和 13。超过一半包含与 ERK 相互作用的 DEF 和/或 D 基序。抑制 ERK 1 小时后,发现 3 种表观遗传调节剂、10 种转录因子、12 种激酶和 1 种 E3 连接酶的调节磷酸化降低。已知与癌症发病机制相关的因子包括 CBX3 S93、MYC S62、FRA1 S265、FOXO3 S294、表皮生长因子受体 (EGFR) T693、RAF1 S29/S642、p70S6KB S423 和 NIPA S344。同样,抑制 ERK 24 小时后,发现 16 种表观遗传调节剂、19 种转录因子、19 种激酶和 8 种 E3 连接酶的调节磷酸化降低。这些磷酸化蛋白质组学结果与对 KRAS 突变型 PDAC 中 ERK 依赖性转录组和蛋白质组的比较结果一致,在其中确定了可归因于 ERK 调节的转录因子的转录特征。此外,他们发现一些 ERK 依赖性蛋白质在蛋白质水平上受到调节,但不在转录水平上受到调节,这表明 E3 连接酶依赖性蛋白质周转。总之,这些结果支持 ERK 依赖性调节复杂的转录组、蛋白质组和磷酸化蛋白质组。
他们利用合成肽库对几乎所有功能性人类丝氨酸-苏氨酸激酶的底物序列特异性进行了体外分析,为每种激酶分配了一个相对激酶活性评分,使用频率因子和显著性来表示激酶基序差异调节的频率(图 4B)。总体而言,参与生长和细胞周期调控的细胞周期依赖性激酶 (CDK)、MAPK、糖原合酶激酶 (GSK) 和 CDK 样激酶 (CLK)(统称为 CMGC 激酶组)失调。抑制 ERK 1 小时后,下调最多的激酶基序是 ERK1 和 ERK2(图4B)。其他下调的激酶包括 ERK 效应激酶、RSK(RSK2、RSK4 和 p70S6KB)以及其他 MAPK(ERK5 和 p38α/β/γ),而在 ERK 抑制 24 小时后,观察到 CDK(CDK1 至 CDK6;CDK1-6)的强烈下调(图 4B)。
利用已建立的激酶-底物网络,他们评估了 ERK 抑制 1 小时和 24 小时后得分最高的相互作用和差异激酶活性(图 4C)。ERK 抑制 1 小时后,下调的激酶网络主要归因于 ERK1/2、RSK2/4 和 p70S6KB。对该网络中归因于 ERK 的磷酸基序进行基序分析,发现原型 [P/L/V/I]-Xp[S/T]-P 基序富集。细胞周期蛋白依赖性激酶 CDK1-6 构成了长时间 ERK 抑制后主要的下调磷酸化网络(图 4C)。在持续抑制 ERK 后,观察到一组ERK 底物的激酶上调,包括 HIPK(HIPK1 至 HIPK3;HIPK1-3)、DYRK(DYRK3 和 DYRK1A 和 DYRK1B;DYRK1A/B)和 CLK(CLK1-CLK4;CLK1/4)激酶(图4B-C)。
然后,他们评估了 KRAS 调节磷酸化蛋白质组中失调的激酶。虽然 KRAS 可以与多种效应子结合,调节不同的信号级联网络,但 ERK 和 KRAS 调节的激酶之间几乎没有差异(图 4D)。值得注意的是,PI3K 效应子 AKT 仅在四种细胞系中的一种 (H358) 中下调。在 KRAS 调节的激酶组中,最一致下调的激酶是细胞周期蛋白依赖性 CDK、ERK MAPK 和 RSK。上调的激酶显示出更多的细胞系异质性,其中 HIPK 和 DYRK 激酶最一致,验证了从 ERK 依赖性激酶组中得出的结论。很少有 KRAS 调节的激酶在与 ERK 相反的方向上显著失调。ERK 活性足以挽救 KRAS 抑制,并且在 KRAS 调节的磷酸蛋白质组中富集。

图4. ERK 通过参与下游激酶来调节动态磷酸化蛋白质组。
(A) 表观遗传调节因子、转录因子、激酶、磷酸酶和 E3 泛素连接酶磷蛋白,其磷酸位点下调,并结合 ERK 的 DEF 和/或 D 基序。(B)分析ERKi 处理 1 小时和 24 小时时 303 S/T 激酶的相对激酶活性。(C) ERKi 处理 1 小时和 24 小时时的激酶-底物网络。(D) 比较了 ERK 依赖性和 RAS 依赖性磷酸蛋白质组中激酶基序富集。
05
ERK 与 RHO 信号高度整合
为了从机制上深入了解 PDAC ERK 磷酸化蛋白质组与 ERK 相比所调控的细胞过程,他们使用GO、KEGG和 Reactome 数据库中的所有基因集进行了通路分析,比较了显著富集的基因集和与每个 ERK 调节的磷蛋白列表相关的所有通路(显著或不显著)的重叠(图 5A)。虽然磷蛋白同一性只有 33%(图 2E),但发现 PDAC ERK 依赖性磷酸化蛋白质组与 ERK 有 66% 的重叠(图 5A)。
本项研究的数据集最富集的是编码与 RHO GTPase 信号通路相关的蛋白质的基因(图 5B),这些基因也是 ERK 中富集最多的基因集之一。在 PDAC ERK 依赖性磷酸化蛋白质组中显著富集但在 ERK 中并不显著的通路与染色质组织、RNA 加工和 G1/S 细胞周期成分有关(图 5B)。因此,他们鉴定出的 ERK 依赖性磷酸化蛋白质组更全面地揭示了异常 ERK 激活在支持 PDAC 的 KRAS 依赖性细胞特性中的作用。
较短的 1 小时 ERK 抑制会改变激酶和底物,这与较长的 24 小时 ERK 抑制所改变的激酶和底物不同。因此,在两个时间点使用 Reactome 基因集评估了下调和上调磷蛋白中的基因集富集情况。ERK 抑制 1 小时后,发现下调磷蛋白中 RHO GTPase 和 EGFR 或 MAPK 相关信号通路富集(图 5C)。上调磷酸位点中没有基因集富集。ERK 抑制 24 小时后,发现下调和上调磷蛋白中存在一组不同的显著富集通路。下调磷蛋白在细胞周期、有丝分裂、染色体维持、DNA 损伤和复制的成分中富集。值得注意的是,与 ERK 抑制 1 小时后 RHO GTPase 磷蛋白富集且磷酸化下调相反,在 24 小时后,这些相同的 RHO 调节通路在上调的磷蛋白中富集。
然后,他们重点研究了 RHO GTPase 相关基因,以检测它们如何从 ERK 抑制 1 小时后在下调的磷酸化蛋白中富集,转变为 ERK 抑制 24 小时后在上调的磷酸化蛋白中富集。他们分析了 RHO GTPase Reactome 通路内蛋白质的磷酸位点及其随时间的变化。在 1 小时后下调的磷酸位点中,很少有在 24 小时后上调的。相反,在每个时间点,上调和下调的磷酸位点都是不同的。然后,他们使用由两个时间点组成的磷酸化网络来分离这些基因集中的蛋白质和相应的激酶(图 5D)。该分析揭示了三个不同的调控网络:第一个由 ERK 底物组成,在 ERK 抑制 1 小时后磷酸位点下调。第二种是 ERK 抑制 24 小时后下调的 CDK1/2/3/5/6 底物,这些底物与 ERK1/2 共有一个底物子集。第三种是一组不同蛋白质底物上的磷酸位点上调,这些磷酸位点是由激酶 PKN1/2、PAK4/6、PDPK1 以及 DYRK、HIPK、CLK 和 SRPK 的假定补偿性激活所驱动的。
RHO 激活的一个研究充分的结果是促进肌动蛋白细胞骨架重组,从而驱动细胞形态的改变。他们使用免疫荧光显微镜评估了 ERK 抑制 24 和 72 小时后肌动蛋白应力纤维的变化。与磷酸化蛋白质组学分析一致,在 ERK 抑制后 24 小时内观察到肌动蛋白应力纤维增加,并且在 72 小时及以后持续增加(图5E-F)。RHO 促进肌动蛋白应力纤维可以激活 YAP1,这是一种经过充分验证的 KRAS 独立性驱动因素。与 RHO 激活作为补偿机制相一致,RHOA 和 YAP1-TEAD 激活已确定为 KRAS G12C突变癌细胞系对adagrasib获得性耐药的基础。

图5. ERK信号与RHO GTPase信号高度整合。
(A) KEGG、GO 和 Reactome 基因集的重叠显著富集(左;P < 0.01)或包含与 ERK 概要以及 ERK 依赖性 PDAC 磷酸蛋白质组中报告的磷蛋白相关的任何基因(右)。 (B) 在 ERK 概要(左)、ERK 依赖性 PDAC 磷酸蛋白质组(中)或 ERK 依赖性 PDAC 磷酸蛋白质组(但未在 ERK 概要中)(右)中显著富集的 Reactome 基因集的通路。 (C) 基因富集分析。(D) 连接Reactome“RHO GTPase 循环”和“RHO GTPase 效应物”基因组内蛋白质的边缘的孤立网络。(E) 免疫荧光的代表性图像。(F) F-肌动蛋白相对整合强度的定量。
06
ERK 调节对 PDAC 生长至关重要的蛋白质
他们注释了 PDAC 中每种具有强遗传依赖性的蛋白质的最高定位分数,并发现核相关磷蛋白富集(76%;288/1072)(图 6A),还发现了相当数量的细胞质蛋白(36%;136/406),这与 ERK 在细胞核和细胞质中调节关键蛋白相一致。
发现 RHO GTPase 相关基因集在 ERK 调节的磷蛋白中富集后,他们接下来评估了 RHO GTPase 磷蛋白的遗传依赖性。在高遗传依赖性中,发现 RHO GTPase 效应子和更集中的 RHOBTB GTPase 通路显著富集。所有高依赖性 ERK 调节的磷蛋白的基因集富集表明 RNA 代谢和细胞周期是最富集的通路,特别是与 M 期和 S 期相关的细胞周期通路(图 6A),这与 ERK 调节细胞周期机制的几乎所有主要成分相一致。
接下来,他们试图探究 ERK 调节的遗传依赖性是否对KRAS突变型癌症具有选择性。由于 90% 以上的 PDAC 病例都含有突变型 KRAS,留下很少的野生型 KRAS PDAC 细胞系可供比较,因此他们扩展了分析范围,以包括DepMap 中 KRAS 突变频率至少占 10% 的其他KRAS突变型癌症。他们将KRAS突变型定义为含有特定的已知 KRAS 驱动突变,包括 G12A/C/D/R/S/V、G13D 或 Q61K/H/L/R。他们比较了含有突变型 KRAS 的细胞系与含有野生型 KRAS 的细胞系中的遗传依赖性。在 KRAS 突变型癌症与 KRAS 野生型癌症中依赖性明显更大的基因中,他们发现 23 个基因的蛋白质产物包括 ERK 调节的磷酸位点(图 6B)。与所有 KRAS 突变癌症相比,这些突变型 KRAS 选择性遗传依赖性在 KRAS 突变型 PDAC 中更为明显(图 6C)。这一结果表明,这些蛋白质虽然对于支持跨癌症类型的增殖很重要,但在 PDAC 中可能更为关键,这可能是因为它对 KRAS 的依赖性更高,并且 ERK 活性更持久。
评估了整个 ERK 调节磷酸化蛋白质组的遗传依赖性后,他们接下来利用激酶-底物网络来归因与高依赖性基因相关的激酶相互作用(图 6D)。这揭示了确定为 ERK1/2 底物的遗传依赖性;下游 ERK1/2 激酶 RSK1、RSK3 和 RSK4 的底物;以及 CDK 底物。他们发现大多数遗传依赖性与下调的磷酸位点有关。在 ERKi 处理 1 小时后,归因于 ERK1/2 磷酸化的最高遗传依赖性是 MYC、ANAPC3 (CDC27)、ANKLE2 和 ANAPC1。值得注意的是,ANKLE2、ANAPC1 和 ANAPC3 是有丝分裂调节剂,而 ANAPC1 和 ANAPC3 是 APC/C 的核心成分,可调节 CDK1 激活剂细胞周期蛋白 B1 和 B2。同时用药理学方法抑制 APC/C 和 ERK 可协同抑制 PDAC 生长。
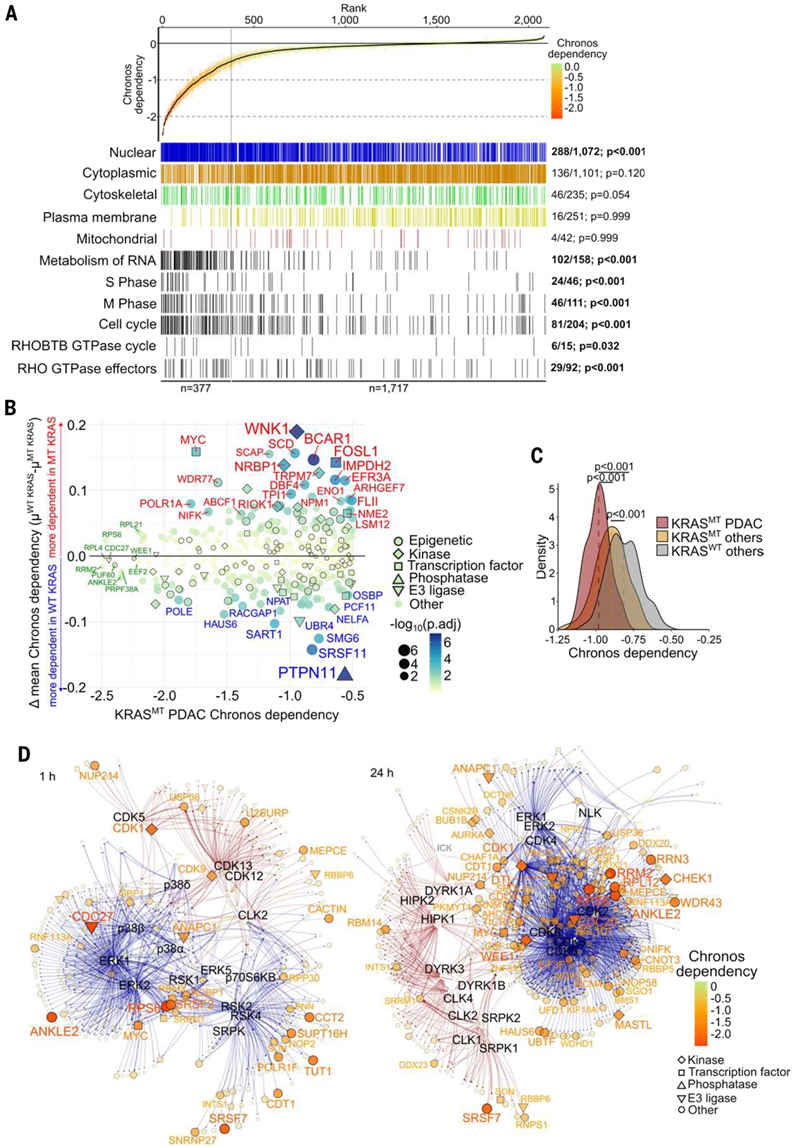
图6. ERK 依赖性 PDAC 磷酸蛋白质组的遗传依赖性。
(A) 带注释的 Chronos 依赖性等级图,按 ERK 调节磷蛋白的 DepMap 依赖性分数排序。(B) ERK 调节磷蛋白对 KRAS 突变型或 KRAS 野生型 (WT) 癌症的遗传依赖性的选择性。(C) 23 个基因的平均遗传依赖性密度直方图。 (D) ) ERKi 处理 1 小时和 24 小时后的激酶-底物网络。
+ + + + + + + + + + +
结 论
本项研究通过分析KRAS 突变型胰腺癌中 ERK 依赖性磷酸化蛋白质组,确定 ERK1 和 ERK2 具有几乎相同的信号传导和转化过程,并且 KRAS 调节的磷酸化蛋白质组几乎完全由 ERK 驱动。本项研究在 2123 种蛋白质上发现了 4666 个 ERK 依赖性磷酸位点,大大扩展了 ERK 依赖性磷酸化事件的深度和广度,并揭示了 ERK 在癌症中更为复杂的功能。本项研究确定 ERK 控制着高度动态和复杂的磷酸化蛋白质组,该蛋白质组集中于细胞周期蛋白依赖性激酶调节和 RAS 同源物鸟苷三磷酸酶功能 (RHO GTPase)。该研究结果建立了最全面的分子图谱和 ERK 驱动 KRAS 依赖性胰腺癌生长的机制。
+ + + + +



