

 English
English文献解读|Protein Cell(21.1):转录组、DNA甲基化组和染色质可及性的综合分析揭示肥厚型心肌病的候选治疗靶点
✦ +
+
论文ID
原名:Integrative analysis of transcriptome, DNA methylome and chromatin accessibility reveals candidate therapeutic targets in hypertrophic cardiomyopathy
译名:转录组、DNA甲基化组和染色质可及性的综合分析揭示肥厚型心肌病的候选治疗靶点
期刊:Protein & Cell
影响因子:21.1
发表时间:2024.05.21
DOI号:10.1093/procel/pwae032
背 景
肥厚性心肌病(HCM)是最常见的遗传性心脏病,其特征是原发性左心室肥厚,通常由肌节基因突变引起,但目前HCM的心脏重塑机制尚不完全清楚。
实验设计
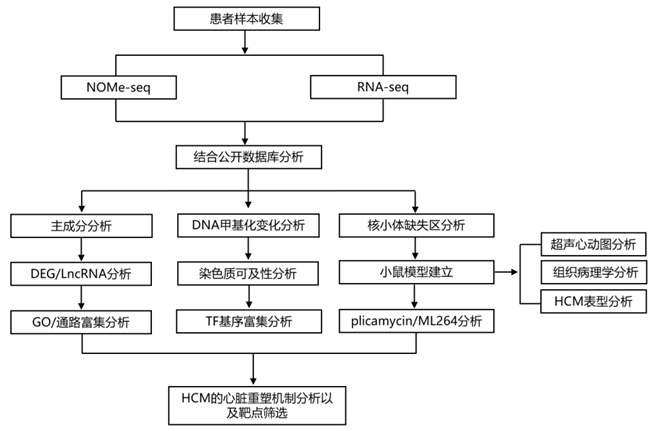
结 果
01

HCM患者与健康供者的转录组比较分析
为探讨HCM的病理机制,研究者团队收集HCM患者心肌进行多组学分析。他们进行了核小体占据和甲基化组测序(NOMe-seq)来分析表观基因组学,包括DNA甲基组和染色质可及性,并进行了转录组分析(RNA-seq)(图1A)。首先,他们研究了HCM的转录组变化。基于蛋白质编码基因的主成分分析(PCA)清晰地将HCM患者与成人对照区分开(图1B)。他们鉴定了两组之间的差异表达基因(DEG)。在HCM患者中,共有691个基因表达上调,这些基因在多种通路中富集,包括细胞外基质组织、细胞外结构组织、细胞粘附和循环系统发育。在HCM患者中,共有835个deg发生下调,这些deg在多种通路富集,包括电子传递链、氧化磷酸化、心脏收缩和代谢过程(图1C)。DEG的上调主要反映了HCM心肌细胞外基质蛋白的激活和过度积累。下调的deg主要参与代谢信号紊乱,这表明HCM是一种心脏能量学改变的疾病。长链非编码rna (lncRNA)也是心脏发育和稳态的重要调节因子。仅使用lncrna的PCA也成功地将HCM患者与成人对照分开(图1D)。在HCM中共鉴定出207个差异表达上调的lncrna和264个差异表达下调的lncrna(图1E)。然后,他们检测了蛋白编码基因与lncrna表达之间的相关性,以确定潜在的顺式调控,发现15对基因上调和15对基因下调(图1F)。在这些基因对中,IDH2在防止小鼠心脏肥大的氧化应激中发挥作用,FHL2可抑制病理性心脏重塑(图1G)。
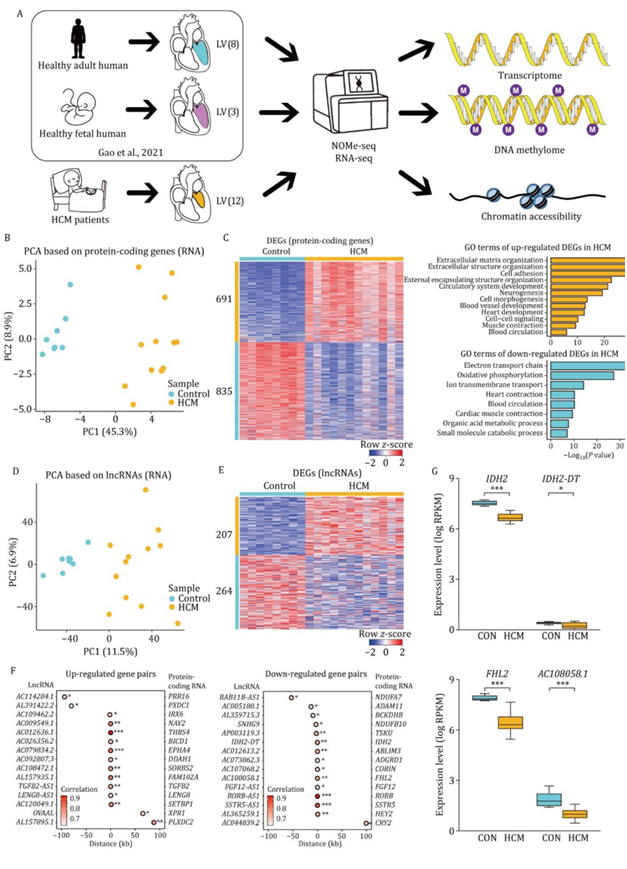
图1. HCM患者和健康成人供体的转录组学分析。
(A) 实验设计示意图。(B) PCA图显示HCM患者和健康供者的蛋白质编码基因的转录组模式。(C) 热图显示HCM患者与健康供者之间蛋白质编码基因(DEG)的表达水平,右侧显示相关的富集通路。 (D) PCA图显示HCM患者和健康供者lncrna的转录组模式。(E) 热图显示HCM患者和健康供者之间DEG的行z分数比例的基因表达水平。(F) 点图显示lncRNA(左)和蛋白质编码基因(右)在顺式调控关系分析中的基因对。(G) 箱线图显示HCM患者和对照组之间具有顺式调控关系的代表性基因对的表达水平。
02
HCM与健康状况的全基因组DNA甲基化分析
接下来,他们研究了HCM心肌中DNA甲基化的变化。HCM患者和对照组的总DNA甲基化水平相似(图2A)。HCM患者和对照组之间基因体周围和侧翼区域的平均DNA甲基化水平分布几乎重叠(图2B)。PCA提示HCM患者与对照组DNA甲基化组存在差异。然后,他们使用甲基化水平差异20%的严格标准确定HCM患者和对照组之间的差异甲基化区域(DMR)。在HCM患者中共鉴定出1453个高甲基化DMR,与这些DMR相关的基因在肌动蛋白丝基过程、血液循环和心脏收缩中富集。HCM患者中共有3600个DMR低甲基化,相关基因在突触组织、细胞粘附和细胞-细胞信号传导中富集(图2C)。高甲基化和低甲基化的DMR都富集在外显子、启动子和增强子区域(图2D)。
在HCM患者和对照组之间,包括外显子、启动子和增强子在内的各种基因组元件的DNA甲基化水平相似。基于启动子、增强子、基因体和CpG岛DNA甲基化的PCA可以区分HCM患者和对照(图2E-F)。HCM患者TPM1启动子的甲基化水平显著升高(图2G-H)。TPM1是一种肉瘤蛋白,TPM1的突变与HCM和先天性心脏缺陷有关。在基因-体区域,HCM患者的TCAP甲基化水平显著升高(图2I-J)。TCAP也是一种肉瘤蛋白,在HCM和扩张型心肌病(DCM)患者中发现其突变。
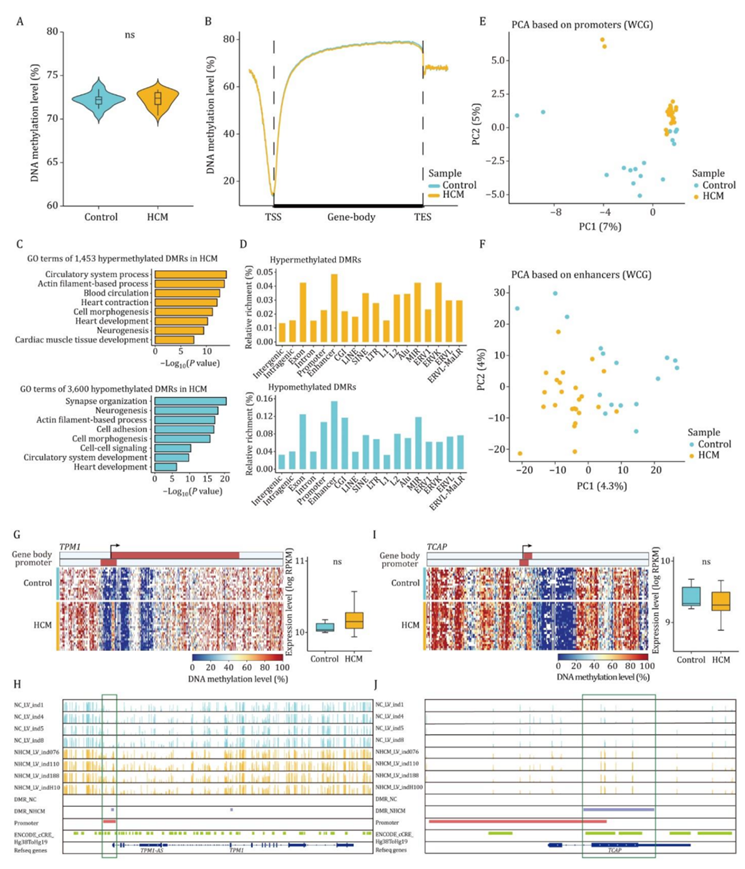
图2. HCM与健康状况的内源性DNA甲基化分析。
(A)小提琴图显示HCM患者和健康供体的全基因组内源性DNA甲基化水平。(B) 线形图显示HCM患者和健康对照者基因体和基因体上游和下游2kb区域的平均内源DNA甲基化水平。(C)GO分析。(D) 条形图显示了位于不同基因组元件的高甲基化和低甲基化DMR的相对富集。(E) PCA图显示HCM患者和健康对照中启动子的内源性DNA甲基化模式。(F) PCA图显示HCM患者和健康对照中增强子的内源性DNA甲基化模式。(G) 热图显示所有HCM患者和对照组中TPM1基因体±10kb区域的内源性DNA甲基化水平。(H) IGV图像显示了4名HCM患者和4名对照组中TPM1及其邻近区域的内源性DNA甲基化水平。(I) 热图显示了基因体±10kb区域的内源性DNA甲基化水平(左),箱线图显示了所有HCM患者和对照组中TCAP的基因表达水平(右)。(J) IGV图像显示了4名HCM患者和4名对照者TCAP及其邻近区域的内源性DNA甲基化水平。
03
HCM心肌染色质可及性的变化
为了绘制HCM的全基因组染色质可及性景观,他们通过胞嘧啶甲基化水平计算了GCH (GCA/GCT/GCC)背景下的染色质可及性。HCM患者与对照组的全基因组水平染色质可及性和转录起始位点(TSS)周围染色质可及性分布相似(图3A-B)。核小体缺失区(ndr)通常含有与活性DNA调控元件相关的转录因子(tf)的结合位点。通过整合HCM患者和健康成人供体的数据,他们确定了30087例近端ndr和484078例远端ndr。与对照组相比,HCM患者近端NDR的染色质可及性水平显著升高。HCM患者远端NDR的染色质可及性水平高于对照组,但差异不显著。PCA表明,HCM患者与对照组的区别在于近端或远端NDR的染色质可及性。他们在HCM患者和健康成人对照的近端和远端ndr中发现了TF结合基序。HCM患者和健康成人对照组共有结合基序,包括STAT(STAT1、STAT3和STAT6)、GATA6和MAZ(图3C)。这些tf的结合基元也在人类和小鼠心脏的四个腔室中富集,表明这些tf具有保守的功能。
基于启动子、基因体和增强子的染色质可及性,PCA明确区分了HCM患者和对照组。NPPA是一个典型的HCM标记基因,几个关键的tf调控NPPA近端启动子。他们发现HCM患者NPPA启动子的染色质可及性水平明显升高(图3D-E)。MYH7编码β-肌球蛋白重链,是HCM中最常见的突变基因之一。HCM患者MYH7基因体的染色质可及性水平高于对照组(图3F-G)。这些HCM关键基因的染色质可及性模式表明,染色质可及性的改变在HCM的病理重塑中起重要作用。
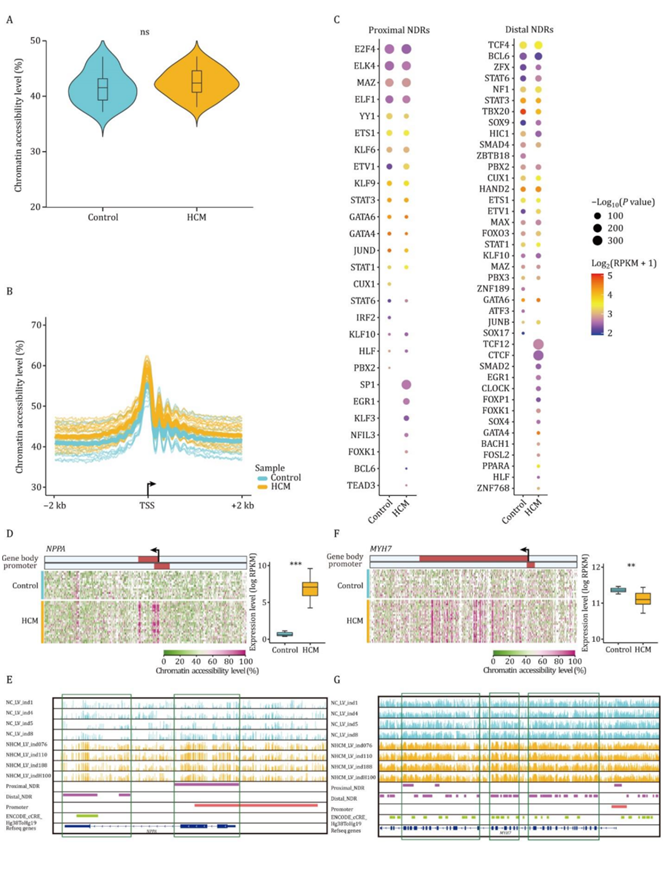
图3. HCM和健康成人对照的染色质可及性谱。
(A) 小提琴图显示HCM患者和健康成人对照全基因组的总可及性水平。(B) 线形图显示HCM患者和对照组TSS上游和下游2kb区域的平均染色质可及性水平。(C) HCM患者和健康成人对照近端(左)和远端(右)NDR的基序富集分析。(D) 热图显示所有HCM患者和对照组NPPA基因体±10kb区域的染色质可及性水平。(E) IGV图像显示了4例HCM患者和4例对照组的NPPA及其邻近区域的染色质可及性水平。(F) 热图显示了基因体±10kb区域的染色质可及性水平(左),箱线图显示了所有HCM患者和对照组中MYH7的基因表达水平(右)。(G) IGV图像显示4名HCM患者和4名对照者MYH7及其邻近区域的染色质可及性水平。
04
HCM心脏在转录组和染色质可及性水平上显示胎儿基因重编程
在各种压力(包括收缩蛋白的基因突变)引发的病理性重塑过程中,成人心脏可能会恢复到胎儿阶段的基因表达谱。在整合公开发表的胎儿数据后,结果显示,关键的肌合成基因,如MYH6、MYH7和ACTC1,在HCM和胎儿心肌中的表达水平低于成人对照组(图4A)。然而,在HCM患者和胎儿中,MYH7/MYH6的比例都增加了,这表明MHC-α(由MYH6编码)和MHC-β(由MYH7编码)异构体开关(图4B)。在HCM患者和胎儿中也检测到能量底物代谢的关键调节因子,如CKM、PDK2和SLC2A4的转录水平降低(图4A)。然后,他们在全转录组水平上探讨了胎儿基因在HCM中的表达模式。PCA清楚地显示PC2轴将HCM和胎儿样本与成人对照分开(图4C)。
与成人对照组相比,297个蛋白质编码基因在胎儿和HCM患者中共同上调,这些基因富集于与细胞外基质组织、心脏发育和肌肉结构发育相关的通路。与成人对照相比,LAMA2、MMP14和POSTN是胎儿和HCM患者中具有代表性的共上调deg(图4D)。据报道,这些基因参与细胞外基质组织等通路,并在HCM中发挥重要作用。在胎儿和HCM患者中共有524个共下调的deg富集于电子传递链和氧化磷酸化等通路,这揭示了HCM患者向胎儿状态的能量代谢转换。本项研究表明,HCM患者倾向于通过下调肌肉合成蛋白和代谢蛋白的表达以及上调细胞外基质基因的表达来恢复胎儿基因谱。
利用基因体或增强子的染色质可及性,HCM患者更接近胎儿,两者都与健康成人对照分开(图4E-F)。然后,通过整合胎儿、成人对照和HCM患者的数据,确定了31092个近端ndr和545960个远端ndr。利用NDR远端染色质可及性的主成分分析也显示HCM患者比成人对照组更接近胎儿,提示HCM中胎儿染色质可及性重编程。他们分析了三组近端和远端NDR中TF结合基序的优先富集模式。几种TF在胎儿和HCM患者(如SP1、EGR1和TCF12)中均富集,或在健康成人对照(如PBX2和SOX17)中均未富集(图4G)。这些结果表明,这些TF可能在病理性心脏重塑中的胎儿基因重编程中发挥作用,并为HCM的新治疗靶点提供了线索。
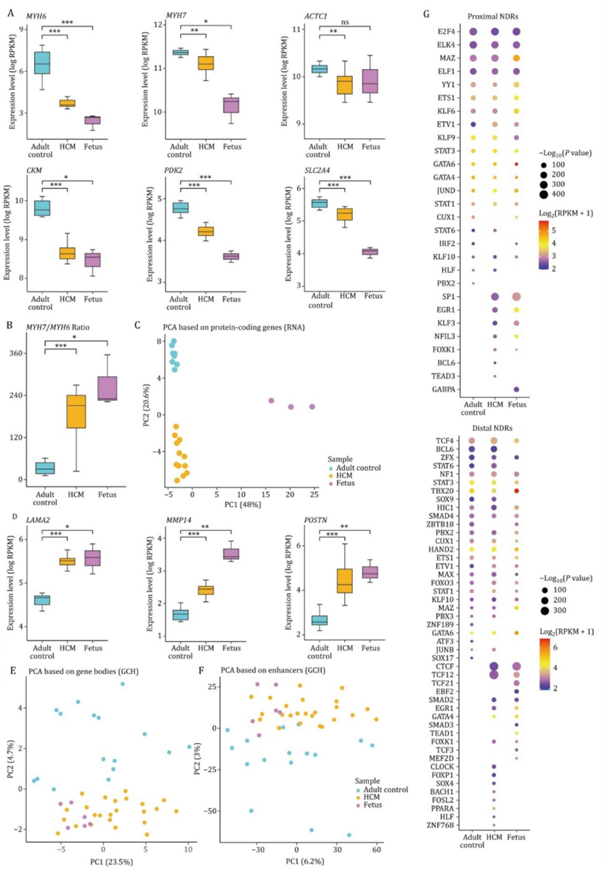
图4. 成人对照、HCM患者和胎儿转录组、DNA甲基化组和染色质可及性的比较。
(A) 箱形图显示MYH6、MHY7、ACTC1、CKM、PDK2和SLC2A4在成人对照、HCM患者和胎儿中的表达水平。(B) 箱线图显示三组MYH7/MYH6比值。(C) PCA图显示了成人对照、HCM患者和胎儿中蛋白质编码基因的转录组模式。(D) 箱形图显示LAMA2、MMP14和POSTN在成人对照、HCM患者和胎儿中的表达水平。(E) PCA图显示成人对照、HCM患者和胎儿基因体的染色质可及性模式。(F) PCA图显示了三组增强子的染色质可及性模式。 (G) 成人对照、HCM患者和胎儿NDR近端(上)和远端(下)基序富集分析。
05
抑制转录因子SP1和EGR1可减轻小鼠HCM
他们建立了一个敲入小鼠模型(Myh6R454C/+Tnnt2R127W/+),具有两种变体,Myh6的p.R454C (NM_010856.4)和Tnnt2的p.R127W (NM_001130179.2),这两种变体分别与人类HCM患者中鉴定的致病突变同源,MYH7的p.R453C和Tnnt2的p.R102W。他们用SP1抑制剂普卡霉素(plicamycin)或EGR1抑制剂ML264处理HCM小鼠,以确定是否可以减轻HCM表型(图5A)。注射前(0 W),超声心动图分析显示,三组HCM小鼠舒张期左室后壁厚度(LVPWd)明显大于野生型(WT)小鼠(图5B)。普卡霉素或ML264处理4周或6周显著减轻了HCM表型,与未处理的HCM小鼠相比,治疗的HCM小鼠的LVPWd厚度显著减少(图5B-C)。与未处理的HCM小鼠相比,用普卡霉素或ML264处理的HCM小鼠心肌细胞横截面面积显著减少(图5D-E),心肌纤维化减少(图5F-G)。这些结果表明,抑制通过增加染色质可及性而激活的tf可以减轻小鼠HCM的发展。
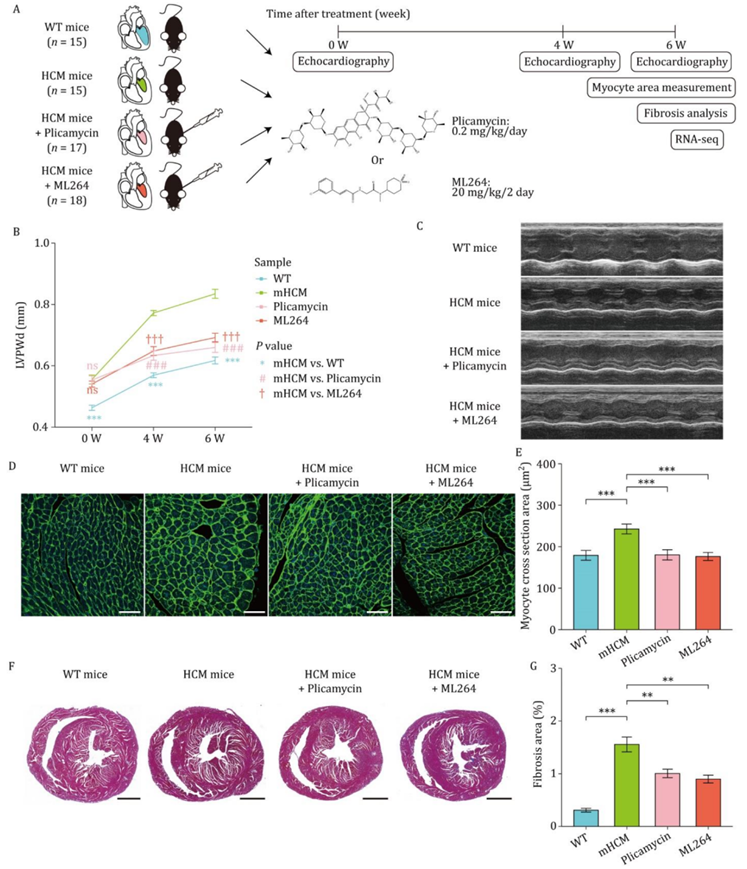
图5. 比较普卡霉素与ML264对WT小鼠、HCM小鼠、HCM小鼠生理病理指标的影响。
(A)显示普卡霉素或ML264注射HCM小鼠的实验过程。(B) 折线图显示了超声心动图对WT小鼠(蓝线)和HCM小鼠LVPWd厚度随时间的变化。(C) 左心室短轴乳头肌切片m型超声心动图的代表性照片。(D) 代表性图像左心室肌切片染色小麦胚芽凝集素(WGA)测量心肌细胞的大小。(E) 条形图显示了基于WGA染色的指示组肌细胞横截面积的定量。(F) 四组心脏切片代表性Masson三色染色图像检测纤维化。
06
普卡霉素和ML264可逆转HCM小鼠胎儿基因重编程
为了探索通过抑制SP1和EGR1(分别使用普卡霉素和ML264)来抑制HCM可能涉及的机制,他们使用心脏组织样本进行了RNA-seq。这些样本来自四组小鼠(野生型小鼠、HCM小鼠、使用普卡霉素或ML264处理的HCM小鼠)。PCA明确区分了未处理的HCM小鼠与其他小鼠(图6A),这表明使用普卡霉素或ML264处理的HCM小鼠的转录组更接近野生型小鼠,而非未处理的突变小鼠。
与野生型小鼠相比,在未处理的HCM小鼠中鉴定出583个上调的DEG,这些基因与mRNA加工、染色质组织和肌动蛋白丝组织有关(图6B)。此外,在HCM小鼠中还鉴定出974个下调的DEG,这些基因主要富集在氧化磷酸化、电子传递链和心脏收缩等通路中(图6C)。与未处理的HCM小鼠相比,在使用普卡霉素和ML264处理的HCM小鼠中分别鉴定出2369和2534个上调的DEG。在这些DEG中,有2101个基因重叠,且富集在氧化磷酸化、电子传递链和心脏收缩等通路中(图6D)。值得注意的是,这些通路与未处理的HCM小鼠相对于野生型小鼠的下调DEG相似(图6C)。与未处理的HCM小鼠相比,在使用普卡霉素或ML264处理的HCM小鼠中分别有1534和1596个DEG下调。在这些DEG中,有1297个基因重叠,这些基因富集在肌动蛋白丝过程、染色体组织和mRNA代谢过程等通路中(图6E)。此外,这些通路与未处理的HCM小鼠相对于野生型小鼠的上调DEG相似(图6B)。
为了研究SP1或EGR1的抑制是否阻止了HCM中的胎儿基因表达重编程,他们首先确定了在HCM小鼠模型中恢复到胎儿表达模式的基因集合。整合了先前研究中小鼠左心室的RNA-seq数据。总共鉴定出88个在胎儿小鼠和HCM小鼠中均上调的DEG;此外,还鉴定出275个共同下调的DEG(图6F)。这363个DEG构成了HCM小鼠中的胎儿基因重编程特征。在这些DEG中,88个共同上调的DEG中有74个和275个共同下调的DEG中有217个(79%)在两种药物处理组中均发生逆转(图6G-H)。这些结果表明,普卡霉素和ML264可以在转录组水平上逆转HCM中的胎儿基因重编程。
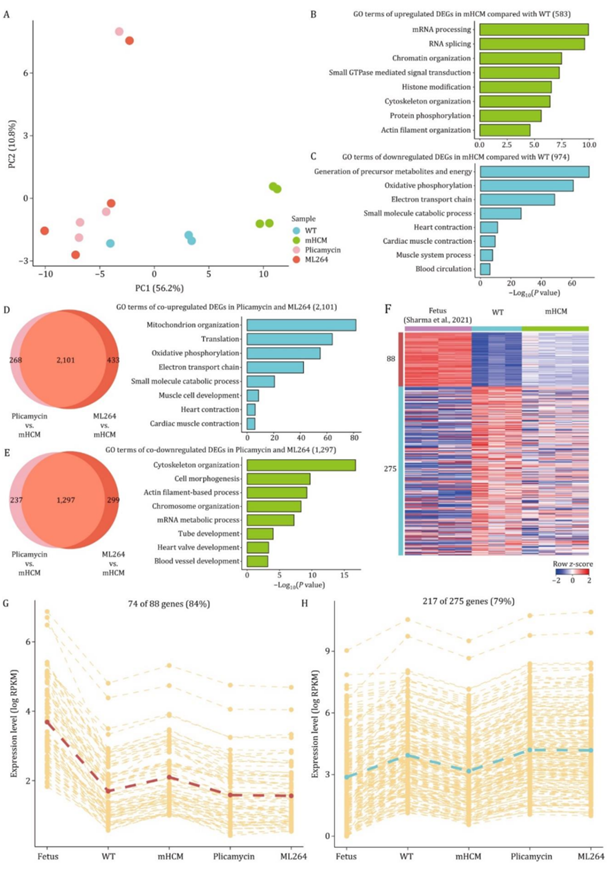
图6. WT小鼠、HCM小鼠、普卡霉素或ML264处理的HCM小鼠和胎鼠的转录组学分析。
(A) PCA图显示了用普卡霉素或ML264处理的WT小鼠、HCM小鼠和HCM小鼠中蛋白质编码基因的转录组模式。(B-C) GO分析。(D) 维恩图显示了普卡霉素或ML264处理的HCM小鼠与HCM小鼠之间重叠上调deg的数量。(E) 维恩图显示了用普卡霉素或ML264处理的HCM小鼠与HCM小鼠之间重叠下调deg的数量。 (F) 热图显示了与WT小鼠相比,HCM小鼠和胎儿中共上调和共下调deg的行z分数刻度基因表达水平。(G) 线形图显示74个基因的表达水平,这些基因在HCM小鼠和胎儿中与WT小鼠相比共上调,而在普卡霉素和ML264处理的HCM小鼠中与HCM小鼠相比共下调。(H) 线形图显示了217个基因的表达水平,这些基因在HCM小鼠和胎儿中与WT小鼠相比共下调,在普卡霉素和ML264处理的HCM小鼠中与HCM小鼠相比共上调。
+ + + + + + + + + + +
结 论
与对照组相比,HCM心肌的转录组、DNA甲基化组和染色质可及性表现出多方面的差异。在转录组水平上,HCM心脏通过减少肌肉和代谢基因表达以及增加细胞外基质基因表达而恢复到胎儿基因程序。在DNA甲基组中,HCM中发现了高甲基化和低甲基化的DMR。在染色质可及性水平上,HCM心脏显示出不同基因组元件的变化。几种TF,包括SP1和EGR1,在HCM的NDR表现出类似胎儿的结合基序模式。特别是,抑制SP1或EGR1在含有肌节突变的HCM小鼠模型中显著减轻突变小鼠的HCM表型并逆转胎儿基因重编程。总的来说,本研究不仅提供了HCM心脏组织的高精度多组学图谱,而且通过干预HCM的胎儿基因重编程,揭示了HCM的治疗策略。
+ + + + +



