

 English
English文献解读|Adv Sci(15.1):KLF5 促进卵巢癌的肿瘤进展和 Parp 抑制剂耐药性
✦ +
+
论文ID
原名:KLF5 Promotes Tumor Progression and Parp Inhibitor Resistance in Ovarian Cancer
译名:KLF5 促进卵巢癌的肿瘤进展和 Parp 抑制剂耐药性
期刊:Advance Science
影响因子:15.1
发表时间:2023.09.13
DOI号:10.1002/advs.202304638
背 景
卵巢癌(OC)是影响女性生殖系统的恶性肿瘤,发病率极高。在妇科肿瘤中,OC 是死亡率最高的肿瘤之一。上皮性卵巢癌 (EOC) 占 OC 病例的 90%,包括高级别浆液性卵巢癌 (HGSOC)、低级别浆液性卵巢癌 (LGSOC)、卵巢子宫内膜样癌 (OEC)、卵巢透明细胞癌(OCCC)和黏液性卵巢癌(MOC) 等亚型。
肿瘤转录组的多样性和复杂性是癌症的主要特征,表观遗传调控元件,如启动子、增强子和超级增强子(SE),在肿瘤中出现特异性或异常激活,通过重塑肿瘤转录组产生肿瘤成瘾或肿瘤特异性转录本 (TST) 来促进肿瘤发生、转移和耐药性。
聚(ADP-核糖)聚合酶 (PARP) 抑制剂 (PARPi) 的开发在治疗患者方面显示出强大的效果。然而,OC是否通过表观遗传调控元件的异常激活来控制转录组重塑并调节PARPi抗性目前尚未有过报道。
实验设计
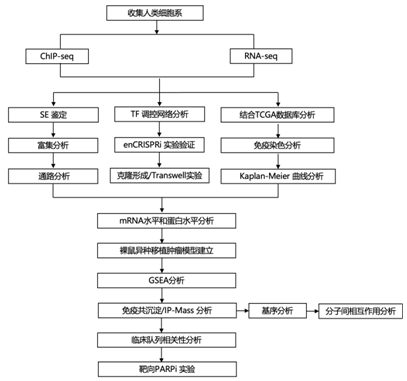
结 果
01

OC 中异常激活的 SE 的情况
为了阐明OC中异常激活的SE元件的特征,研究者团队对6个OC细胞(OVCA420、OVCA429、OVCAR3、TOV21G、ES2和A2780)和1个正常卵巢上皮细胞(IOSE80)中的H3K4me1、H3K4me3、H3K27Ac、polII和EP300,进行了染色质免疫沉淀测序(ChIP-seq),并对OVCA420细胞的BRD4进行了ChIP-seq测序(图1A)。他们对复旦大学上海癌症中心(FUSCC)的卵巢癌进行了RNA-seq数据分析,并从ENCODE网站上收集了6个卵巢癌细胞系(NCI_ADR-RES、IGROV1、OVCAR4、OVCAR5、OVCAR8和SKOV3)的公开H3K27Ac ChIP-seq数据。
他们重点关注富含H3K27Ac信号且远离基因转录起始位点(TSS)的区域(TSS上游/下游2.5 kb),因为这些区域在以往的研究中属于增强子区域。他们在 OC 中识别出 2481 个异常激活的 SE 元件,包括 HGSOC 中的 1651 个、OCCC 中的 1038 个和 OEC 中的 744 个(图 1B)。
他们选择了三种 OC 亚型的代表性细胞系来研究可能受 SE 调控的邻近基因。例如,EGFR和KLF5在HGSOC细胞中富集(图 1C);REST和MET在OEC细胞中富集(图 1D);CDKN2A和CDK6在OCCC细胞中富集(图 1E)。他们还比较了SE在三种细胞系亚型中出现的程度,发现属于同一亚型的细胞系具有更高程度的SE相似性(图 1F)。
为了揭示不同OC亚型中SEs相关的关键信号通路,分析了SE富集的邻近基因,发现OEC亚型富集了铁死亡、NF-κB等信号通路;HGSOC亚型在趋化性和胆碱代谢信号通路方面丰富;OCCC亚型主要富集TGF-β和Wnt信号通路。所有三种亚型均富含 Hippo、MAPK 和转录异常信号通路(图 1G)。总的来说,这些结果提供了对 OC 中异常激活的 SE 及其在不同 OC 亚型中的存在和特征的全面了解。

图1. 描述OC中的超级增强元件。
(A) 用于识别 OC 中异常激活的 SE 的分析策略的流程图。(B) 维恩图显示了每个 OC 亚型中 SE 的数量。 (C-E) OVCAR5中SE的数量、igrpv1、ES2细胞和相邻代表性基因的表示。(F) 矩阵显示了在不同细胞类型中检测到的 SE 的成对相似性。(G) 三种 OC 亚型中 SE 邻近基因富集的特定和共享的 KEGG 信号通路。
02
SE驱动KLF5转录和KLF5自转录调节
由于转录因子 (TF) 参与SE的调控网络,他们分析了来自FUSCC的30个OC和8个正常卵巢RNA测序数据中差异表达的TF,其中在这些TF的基因附近发现了异常激活的超级增强子元件。火山图表明KLF5、PAX8、SOX17、ELF3和HOXB3在 OC 组织中高度表达(图 2A)。此外,他们构建了这些 TF 的潜在下游靶基因的调控网络(图 2B)。他们在OVCA420和OVCAR5 OC细胞中发现KLF5启动子上游的两个超增强子调控元件发生激活(图2C)。在OVCA420细胞中,两个SE显示H3K4me1和H3K27Ac的组蛋白修饰信号,与EP300和BRD4结合,伴随H3K4me3的低修饰信号水平KLF5和BRD4蛋白结合发生在它们的启动子区域,这表明KLF5可能受到 SE 的转录调节和自我调节。
为了检验这一假设,他们首先使用 enCRISPRi 系统抑制这两个超级增强子的活性,并发现单独或联合抑制两个 SE 的活性,能显著下调H3K27Ac 水平和KLF5 mRNA 水平(图 2D)。一致地,抑制两种SE活性消除了 OVCA420细胞的克隆形成(图2E)和侵袭能力(图 2F)。
他们通过ChIP-qPCR实验证实了KLF5和BRD4在KLF5启动子区域的结合(图2G),并在OVCA420细胞中设计了靶向KLF5和BRD4的小干扰RNA (siRNA),敲低KLF5或BRD4显著抑制KLF5启动子活性(图2H)。BRD4敲低显著降低了KLF5 mRNA(图 2I)和蛋白质水平(图 2J)。这些结果表明,SE 的异常激活调节KLF5和KLF5的转录进一步结合其自身的启动子区域以维持自身高转录表达。

图2. SE 在 OC 中驱动KLF5转录和KLF5自转录调节。
(A) 使用 FUSCC 队列 RNA-seq 数据,火山图显示 OC 中 TF 的差异表达,包括 8 个正常卵巢组织和 30 个 OC 组织。(B) TF 的差异表达及其下游基因的调控网络。(C) H3K4me1、H3K4me3、H3K27Ac、EP300、KLF5和BRD4在KLF5启动子和两个上游 SE 处的 ChIP-seq。 (D) 转染 dCas9-KRAB、靶向KLF5 SE 的 sgRNA 和 MCP-LSD1 慢病毒的 OVCA420 细胞中KLF5的 mRNA 水平。(E-F) 使用 enCRISPRi 慢病毒阻断KLF5 SE 可抑制 OVCA420 细胞中的集落数量形成和迁移能力 (G) ChIP-qPCR 测定显示OVCA420 细胞中KLF5和BRD4在KLF5启动子处的结合。 (H) OVCA420和OVCAR3细胞中用KLF5或BRD4 siRNA转染的KLF5启动子的荧光素酶活性。(I-J) BRD4 sirna转染的KLF5在OVCA420和OVCAR3细胞中的mRNA水平和蛋白水平。
03
KLF5的高表达是OC特有的并与不良预后相关
为了阐明KLF5的特征,他们从癌症基因组图谱 (TCGA) 公共数据库中收集了 21 种正常组织和癌症组织的 RNA-seq 数据。他们发现KLF5在OC、胆管癌、胃癌等肿瘤中高表达。其中,KLF5表达的差异在OC中尤其显著(图 3A)。图2A中 FUSCC 的 RNA-seq 数据也突出显示了KLF5的过度表达,表明KLF5可能是OC中相对特定的驱动因子。在 FUSCC 队列 1 的 OC 组织中发现了 类似的KLF5高表达模式(图3B),KLF5高表达的OC患者总生存率和无病生存率较低(图3C-D)。因此,他们验证了KLF5FUSCC队列2(包括74例正常卵巢组织和165例OC组织)的蛋白质水平,发现KLF5的蛋白水平在OC组织中也高表达(图 3E-F)。KLF5蛋白水平高的 OC 患者的总生存率(图 3G)和无病生存率(图 3H)也较低。

图3. KLF5在 OC 中特异性过度表达,并且在临床上与患者预后相关。
(A) TCGA 数据集中不同正常和肿瘤样本中KLF5的 mRNA 水平。 (B) FUSCC 队列 1 中正常和 OC 样本中KLF5的 mRNA 水平。(C-D) FUSCC 队列 1 中总生存和无病生存的 Kaplan-Meier 曲线。(E) FUSCC 队列 2 的 74 个正常组织和 165 个 OC 组织中KLF5的代表性免疫染色图像。(F) 正常组织和 OC 组织中KLF5蛋白水平的免疫印迹。 (G-H) 用KLF5蛋白水平分层的FUSCC队列2的总生存期和无病生存期的Kaplan-Meier曲线。
04
KLF5驱动OC的生长和转移
由于KLF5在 OC 中高表达,他们接下来探究了KLF5在 OC 中的致癌功能,设计了两种针对KLF5 的siRNA ,发现KLF5的敲低显著抑制了OVCA420 和 TOV21G OC 细胞中的克隆形成(图 4A)和迁移(图 4B)。KLF5高表达与细胞增殖之间存在显著的正相关性,表明KLF5在促进 OC 细胞增殖过程中发挥着至关重要的作用(图 4C)。然后他们构建了两个针对KLF5的guide(g) RNA质粒,将其与CRISPR质粒一起包装成慢病毒,并感染高表达KLF5的OVCA420和SKOV3细胞(图 4D)。CCK-8、克隆形成和 Transwell 检测表明KLF5敲低显著抑制增殖(图 4E-F)和OC 细胞的迁移(图4G) 。
为了验证在体内的发现,他们用Cas9和对照gRNA敲低慢病毒感染SKOV3细胞,并皮下注射到6周龄裸鼠体内。结果表明,KLF5基因敲除可显著降低OC细胞的肿瘤大小(图 4H-I)。用Cas9和对照gRNA或sgKLF5敲低慢病毒感染TOV21G细胞,并腹腔注射裸鼠。PET-CT结果显示,OC细胞中KLF5敲低可减少SUV摄取(图4J-K)和并减少腹部肿瘤结节数量(图4L)。

图4. KLF5驱动 OC 肿瘤生长和转移。
(A-B) 用KLF5 siRNA 或对照 siRNA转染的 OVCA420 和 TOV21G 细胞的克隆形成测定和迁移测定。 (C) Depmap 数据集中 OC 细胞系中KLF5 mRNA 水平与 CRISPR 影响值(增殖能力)之间的相关性。(D) 感染 Cas9 和KLF5 sgRNA 的 OVCA420 和 SKOV3 细胞中KLF5蛋白水平的免疫印迹。 (E) CCK8测定。 (F-G)克隆形成实验和迁移实验。(H) 裸鼠的异种移植瘤。 (I) KLF5的敲除减少了异种移植肿瘤的重量。(J) PET-CT显示感染Cas9和KLF5 sgRNA的TOV21G细胞的腹部肿瘤转移。(K-L) 感染Cas9和KLF5 sgRNA的TOV21G细胞腹膜腔的相对SUV最大值和结节数。
05
KLF5调控RAD51转录和HRR通路
为了进一步研究KLF5促进 OC 进展的分子机制,他们分析了对照和KLF5敲除 OVCA420 细胞的 RNA-seq 基因表达谱。GSEA显示KLF5敲低细胞中下调的前5个信号通路富集于DNA复制、错配修复、剪接体、HR和细胞周期(图 5A-B)。
由于KLF5作为转录因子通过控制与特定基因组区域的结合并调节靶基因转录来发挥生物学功能,因此他们在 OVCA420 细胞中对KLF5进行了 ChIP-seq 。结果表明获得了2497个KLF5结合位点,主要位于基因启动子区域(图 5C-D)。接下来,他们分析了 ChIP-seq 和 RNA-seq 数据,发现 32 个靶基因可能受KLF5转录调控(图 5D)。使用OVCA420细胞的CRISPR/Cas9功能筛选数据,发现32个基因中有18个与细胞增殖相关(图 5E)。
由于KLF5的敲低显著抑制了OC OVCA420细胞中的HRR通路,并且对这18个基因的功能测序和分析表明它们含有核心同源重组基因RAD51 ,他们推测KLF5可能重塑RAD51转录。为了检验这一假设,首先观察了RAD51基因组区域中的KLF5结合位点,并发现RAD51启动子中显著富集(图 5F)。
KLF5敲低显著降低了OC 细胞中RAD51 mRNA 水平(图S5B,支持信息)和蛋白质水平(图 5G ),并增加了磷酸化 γ-H2AX 的蛋白质水平。此外,KLF5敲低促进了OVCA420和TOV21G OC细胞中的DNA损伤(图 5H)。免疫荧光染色显示,抑制KLF5会减少 RAD51灶点并增加 γ-H2AX 灶点(图 5I-K),表明KLF5在调节卵巢癌细胞的 HRR 中发挥着至关重要的作用。
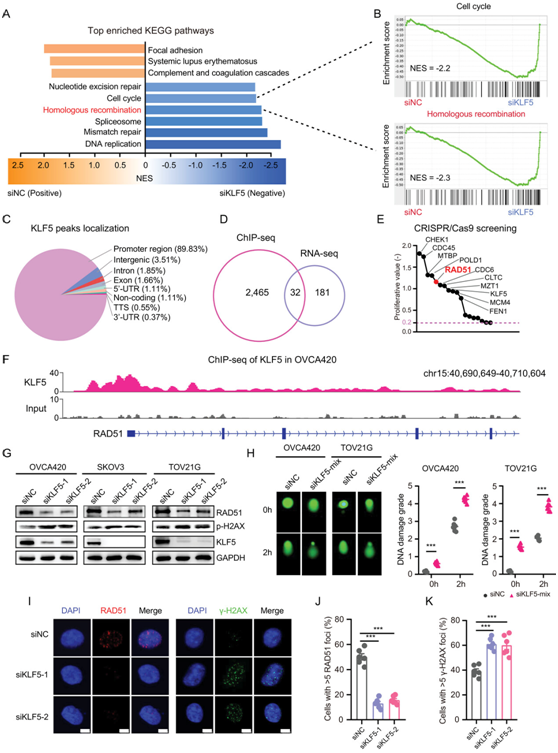
图5. KLF5调节RAD51转录和 HRR 通路。
(A) 在用KLF5 siRNA 或对照 siRNA转染的 OVCA420 细胞中,前六个富集的 KEGG 下调通路和三个上调的通路。(B) GSEA富集了转染KLF5 siRNA或对照siRNA的OVCA420细胞的细胞周期和同源重组通路。(C) KLF5在OVCA420细胞中的全基因组结合位点,大部分位于启动子区域。(D) KLF5 ChIP-seq和RNA-seq数据的韦恩图显示了32个在OVCA420细胞中受KLF5转录调控的基因。 (E) Depmap 数据集中的 OVCA420 中的KLF5靶基因表明这些基因参与了 OC 细胞增殖。(F) OVCA420 中KLF5的 ChIP-seq 数据显示KLF5在RAD51启动子区域的结合。 (G) 用KLF5 siRNA或对照siRNA转染的OVCA420、SKOV3和TOV21G细胞中RAD51、p-H2AX、KLF5蛋白水平的免疫印迹。 (H) 用KLF5 siRNA 或对照 siRNA转染的 OVCA420 和 TOV21G 细胞中的彗星测定。(I)免疫荧光染色。(J-K) RAD51灶和γ-H2AX灶的定量结果。
06
KLF5与EHF、ELF3形成转录复合物
为了更好地了解KLF5对RAD51的特异性转录调控机制,他们首先使用KLF5靶向抗体进行蛋白质免疫沉淀,然后进行质谱 (IP-Mass)分析,发现了48个潜在的与KLF5相互作用的蛋白质,其中包括9个核蛋白(图 6A )。对九种蛋白质的结合基序分析表明,EHF和ELF3在基因组中可能与KLF5重叠(图6B)。同样,在OVCA420和OVCAR3细胞中,通过CO-IP可以很容易地检测到KLF5、EHF和ELF3之间的相互作用(图6C)。
为了阐明EHF和ELF3与RAD51启动子的结合能力,他们构建了PCDH-3X-Flag- EHF和PCDH-3X-Flag- ELF3过表达质粒,将其包装成慢病毒,并感染OVCA420细胞进行ChIP-seq。数据显示EHF和ELF3也在RAD51中富集启动子区域,该区域也与KLF5结合(图 6D)。重要的是,OVCA420和OVCAR3 OC细胞中EHF和ELF3的敲低削弱了polII与RAD51启动子的结合(图 6E),并降低了RAD51启动子活性,最终导致RAD51 mRNA下调(图 6F)和蛋白质水平降低(图 6G)。
为了进一步确定该蛋白质转录复合物中的关键调控因子,他们将靶向KLF5的 siRNA 转染至 OVCA420 和 OVCAR3 细胞中,发现KLF5敲低抑制了EHF和ELF3蛋白水平,表明KLF5可能是最关键的调节蛋白(图 6H)。此外,他们观察到KLF5与ELF3启动子以及上游SE区和EHF启动子区结合(图6I),表明KLF5也可能调节EHF和ELF3的转录。因此,他们将靶向KLF5的siRNA转染到OVCA420和OVCAR3细胞中,发现KLF5敲低也抑制了EHF和ELF3的mRNA水平(图6J-K)以及抑制了EHF和ELF3对RAD51启动子的结合能力(图6L)。

图6. KLF5与EHF和ELF3形成转录复合物。
(A) 免疫沉淀-质谱法。(B) OVCA420 细胞中KLF5、EHF和ELF3的基序分析。 (C) 免疫共沉淀分析显示OVCA420 和 OVCAR3 细胞中内源性KLF5、EHF和ELF3之间的相互作用。(D) ChIP-seq 数据显示EHF、ELF3和KLF5在RAD51中富集OVCA420 细胞中的启动子区域。(E) 在转染EHF、ELF3 siRNA或对照siRNA的OVCA420和OVCAR3细胞中,polII在RAD51启动子区域的富集能力。 (F-G) 用EHF、ELF3 siRNA或对照siRNA转染的OVCA420和OVCAR3细胞中的RAD51 mRNA水平和蛋白质水平。 (H) 用KLF5 siRNA或对照siRNA转染的OVCA420和OVCAR3细胞中的EHF、ELF3和KLF5蛋白水平的免疫印迹。 (I) KLF5、H3K4me1、H3K27Ac、H3K4me3和polII的ChIP-seq数据显示,在OVCA420细胞中,KLF5富集于ELF3启动子区、SE区和EHF启动子区。(J-K) 用KLF5 siRNA 或对照 siRNA转染的 OVCA420 (J) 和 OVCAR3 (K) 细胞中的EHF和ELF3 mRNA 水平。 (L) 用KLF5 siRNA或对照siRNA转染的OVCA420细胞和OVCAR3细胞中RAD51启动子区域中EHF和ELF3的富集能力。
07
EHF和ELF3高表达并与不良预后相关
KLF5在促进 OC 癌发生中具有关键作用,但对于EHF和ELF3在 OC 患者中的临床意义和分子功能知之甚少。RT-qPCR 验证EHF mRNA 在 FUSCC 队列 1 的 OC 组织中显著过表达(图 7A)。然而,在 OC 患者中,EHF的 EHF 表达与总生存期(图 7B)或无病生存期(图 7C)之间没有显著相关性。ELF3的 mRNA 水平在 FUSCC 队列 1 的 OC 样本中高表达(图 7D),而ELF3 高表达的 OC 患者表现出较低的总生存率(图 7E)和无病生存率(图 7F)。免疫组化(IHC)结果显示,与 FUSCC 队列 2 中的正常组织相比,OC 组织中EHF(图 7G)和ELF3(图 7H )蛋白过度表达。
EHF蛋白高表达的 OC 患者表现出较低的总体生存率(图 7I),但与无病生存率没有显著相关性(图 7J)。ELF3蛋白高表达的OC患者表现出较低的总生存率(图 7K)和无病生存率(图 7L)。
TCGA 和 GSE2109 数据集还揭示了 OC 样本中它们的 mRNA 水平之间存在显著的正相关性。更重要的是, KLF5、EHF和ELF3蛋白水平较高的 OC 患者表现出较低的总生存率(图 7M)和无病生存率(图 7N)。这些数据表明KLF5联合EHF和ELF3可能是OC患者的有效预后因素。

图7. EHF和ELF3在 OC 患者中的临床意义。
(A) FUSCC 队列 1 中正常样本和 OC 样本中的 EHF mRNA 水平。(B-C) FUSCC 队列 1 中总生存期和无病生存期的 Kaplan-Meier 曲线。(D) FUSCC 队列 1 中正常和 OC 样本中ELF3的 mRNA 水平。 (E-F) FUSCC 队列 1 中总生存和无病生存的 Kaplan-Meier 曲线。(G-H)免疫染色图像。 (I-N) Kaplan-Meier 曲线。
08
靶向KLF5增加 OC 患者对 PARPi 耐药的敏感性
鉴于HRR是肿瘤中PARPi耐药的核心途径,并且已证明KLF5通过重塑OC中的RAD51转录来调节HRR,他们假设KLF5可能参与OC细胞中的PARPi耐药,首先验证了OC细胞系中的KLF5蛋白水平,发现KLF5在OVCA420、SKOV3、TOV21G等细胞系中高表达(图 8A)。
有趣的是, KLF5蛋白水平与临床治疗中广泛使用的PARP抑制剂olaparib的IC50值之间存在显著的正相关性(图 8B)。类似高KLF5与四个olaparib敏感的异种移植瘤(PDX) 样本相比,四个olaparib耐药的患者来源的 OC 的PDX中发现了蛋白质表达水平(图 8C)。此外, KLF5上游2号SE在olaparib抗性PDX样品中显示出更高的修饰H3K27Ac信号(图 8D)。
他们进一步构建了olaparib耐药细胞系,以确认KLF5的SE是否驱动KLF5表达、增强RAD51转录并导致PARPi耐药。数据显示,olaparib耐药的 ES2 和 A2780 细胞中KLF5和RAD51 mRNA 水平升高(图 8E),并且KLF5、RAD51和BRD4存在类似的蛋白表达模式(图 8E)。
重要的是,olaparib诱导SE 2 号元件上游 KLF5的激活(图8F)。通过靶向BRD4 siRNA抑制KLF5的 SE 活性可降低KLF5 mRNA 水平,表明 PARPi 诱导KLF5的 SE 激活并驱动OC 细胞中的KLF5过度表达。
组蛋白脱乙酰酶抑制剂辛二酰苯胺异羟肟酸 (SAHA) 通过增加KLF5蛋白赖氨酸369 (K369) 乙酰化水平来抑制乳腺癌中KLF5蛋白的表达。因此,他们探究了SAHA对OC细胞中KLF5表达的调节作用。SAHA处理显著增加了SKOV3、TOV21G和OVCA420细胞中 KLF5的乙酰化水平,并抑制了KLF5和RAD51的总蛋白水平(图8G)。令人惊讶的是,SAHA联合olaparib显著抑制OVCA420和SKOV3细胞的增殖,而OVCA420和SKOV3细胞对olaparib不敏感(图8H)。此外,SAHA显著提高了SKOV3和TOV21G 细胞对olaparib的敏感性(图8I),提示SAHA在PARPi耐药和KLF5高表达的OC患者的治疗中具有潜在的作用。为了验证这一功能,他们构建了KLF5高表达的olaparib耐药PDX小鼠模型,并发现与SAHA或olaparib处理相比,olaparib联合SAHA显著抑制皮下肿瘤形成(图8J-K)。
他们还将KLF5高表达的对olaparib不敏感的OC细胞TOV21G注射到SAHA和olaparib处理的小鼠腹腔内,发现与单独SAHA或olaparib处理相比,olaparib联合SAHA显著抑制了TOV21G细胞的生长和腹腔转移(图8L-N)。
总之,这些数据表明 PARPi 耐药 OC 患者表现出KLF5 SE 的异常激活并促进KLF5高表达。KLF5通过重塑RAD51转录和增强 HRR 途径来操纵 OC 中的olaparib耐药性。SAHA联合olaparib可能是针对KLF5高表达水平的PARPi耐药OC患者的潜在治疗策略(图 8O)。

图8. KLF5调控 OC 对 PARP 抑制剂的敏感性。
(A) 11 种 OC 细胞系中KLF5蛋白水平的免疫印迹。 (B) OC细胞系中KLF5蛋白水平与olaparibIC50值之间的相关性。 (C) OC olaparib敏感 (Ola-S) 或耐药 (Ola-R) PDX 样本中KLF5蛋白水平的免疫印迹。(D) 通过 H3K27Ac 信号的 ChIP-qPCR 测定olaparib敏感或耐药的 OC PDX 样品中KLF5超级增强子 2 的活性。(E) ES2、A2780 和olaparib处理的 ES2 和 A2780 细胞中的KLF5 mRNA 水平(左)和KLF5、RAD51、BRD4蛋白水平(右)。 (F) KLF5超级增强子2的活性通过 H3K27Ac 信号的 ChIP-qPCR 测定 ES2、A2780 和olaparib处理的 ES2 和 A2780 细胞中的 H3K27Ac 信号。 (G) 用 DMSO 或 SAHA (1µm)处理的 SKOV3、TOV21G 和 OVCA420 细胞中KLF5和RAD51蛋白水平的免疫印迹。(H) 克隆形成实验结果。(I) olaparib或olaparib联合SAHA处理SKOV3和TOV21G细胞的IC50值。(J-K) 异种移植物肿瘤和肿瘤体积。 (L-N) 荧光素酶图像、荧光素酶值、腹腔结节数显示在BALB/ c裸鼠中,DMSO、olaparib、SAHA、olaparib联合SAHA处理TOV21G细胞的腹腔肿瘤转移。(O) SE驱动KLF5的示意图。
+ + + + + + + + + + +
结 论
本项研究鉴定了在 OC 中异常激活的超级增强子调节元件,并发现 SE 驱动 OC 患者中转录因子 KLF5 和PARPi的相对特异性表达。KLF5表达与 OC 患者的不良预后相关,并且可以在体外和体内驱动肿瘤进展。从机制上讲,KLF5与EHF形成转录复合物和ELF3并与RAD51的启动子区域结合以增强其转录,从而加强同源重组修复 (HRR) 通路。值得注意的是,SAHA和olaparib的联合处理显著抑制具有高KLF5的PARPi耐药OC细胞的肿瘤生长和转移。总之,本项研究发现 SE 驱动的KLF5是 OC 进展和 PARPi 耐药的关键调节因子,并确定了针对 PARPi 耐药和高KLF5的 OC 患者的潜在治疗策略。
+ + + + +



