

 English
English文献解读|Cell Rep Med(14.3):男男性行为者肠道微生物群的建立与性行为有关
✦ +
+
论文ID
原名:Establishment of a non-Westernized gut microbiota in men who have sex with men is associated with sexual practices
译名:男男性行为者肠道微生物群的建立与性行为有关
期刊:Cell Reports Medicine
影响因子:14.3
发表时间:2024.02.15
DOI号:10.1016/j.xcrm.2024.101426
背 景
男男性行为者 (MSM) 的肠道微生物群与非 MSM 的肠道微生物群不同。人类肠道微生物群受到多种因素的影响,包括健康状况和环境条件,但个体间的巨大差异仍然无法解释。
实验设计
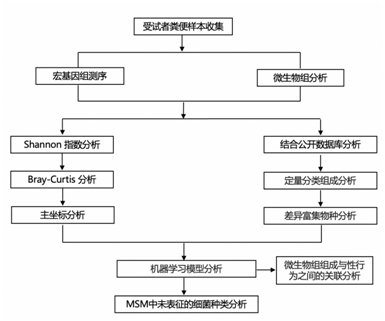
结 果
01

MSM 在物种水平上增加了多样性并改变了肠道微生物组结构
研究者团队对总共 124 名受试者(MSM n = 93,非 MSM n = 31)的粪便样本进行了 宏基因组测序,使用基于MetaPhlAn 4分析具有相对丰度定量的微生物群落组成(图S1A)。在 124 名受试者中,他们总共鉴定出 1675 个 物种级基因组箱 (SGB),其中包括 1124 个已知 SGB (kSGB) 和 551 个未知SGB (uSGB)。
使用 Shannon 指数比较 α 多样性显示,MSM 中的微生物组多样性高于非 MSM(图1A)。为了研究 MSM 和非 MSM 微生物组的差异,他们使用 Bray-Curtis 距离计算了 β 多样性,执行控制混杂因素(年龄、种族、抗生素使用、BMI 和 HIV 感染)的排列多元方差分析 (PERMANOVA),并证实了 MSM 和非 MSM 之间的物种水平微生物组差异(图1B)。根据主要协变量组(例如年龄、BMI、HIV 状况或最近的抗生素摄入量)对受试者进行分层的附加分析验证了观察结果(图 S1 B)。检查计数等免疫学参数的影响对 103 名 HIV 感染者中CD4+ T 细胞的频率和频率以及 CD4/CD8 T 细胞比率进一步证实,肠道微生物组变化与 MSM 之间存在显著关联,与 HIV 感染无关(图 S1 C)。
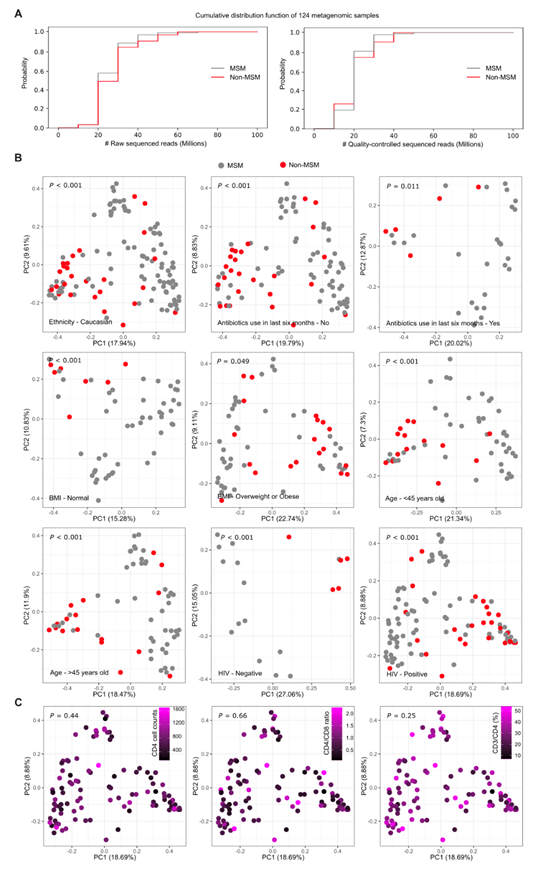
图S1. 测序读取数的基本统计,以及男男性行为人群样本与非男男性行为人群样本在人体测量因素和HIV感染方面分类组成的比较。
(A) MSM和非MSM样本中测序reads的累积分布函数。(B) 主坐标分析(PCoA)。(C) 基于HIV感染个体的Metaphan 4丰度谱的PCoA分析。
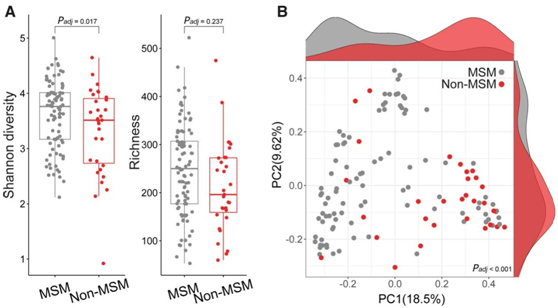
图1. MSM 和非 MSM 的肠道微生物群落结构。
(A) MSM 与非 MSM 相比的 Alpha 多样性分析。 (B)在 MSM 和非 MSM 样品之间分析的微生物丰度的主坐标分析 (PCoA)。
02
在MSM中以普雷沃氏菌(Prevotellaceae)为主的群落包括Segatella copri复合体成员和未知物种
他们比较了MSM 和非 MSM 之间普雷沃氏菌属所有物种的丰富度、丰度和流行率。值得注意的是,MSM 微生物组中普雷沃氏菌物种的平均数量是非 MSM 微生物组的三倍(图2A)。为了比较普雷沃氏菌群落多样性,他们专门分析了普雷沃氏菌成员的香农指数,证实了与非 MSM 相比,MSM 肠道微生物组具有更高的 α 多样性(图2A)。基于以普雷沃氏菌为中心的β多样性分析,他们还观察到MSM和非MSM样本之间存在显著的分离(图2B)。在本研究鉴定的47种普雷沃氏菌细菌(kSGB n= 28, uSGB n= 19)中,仅考虑在所有样本的90th百分位上相对丰度最低为0.1的物种时,有16种在MSM中存在差异富集(图2C)。
未知的普雷沃氏菌成员 (uSGB) 在 MSM 肠道微生物组中的比例同样过高,与非 MSM 相比,其丰度和患病率较高(图 2C)。属于S. copri物种复合体的成员是MSM中最常见和最丰富的普雷沃氏菌科成员,70%的MSM具有至少两个S. copri分支(图2C-D)。此外,在典型的非西化(Non-Wes)微生物组中观察到的来自多个S. copri分支的菌株的共存特征,在男男性行为人群中始终高于非男男性行为人群(图2D)。
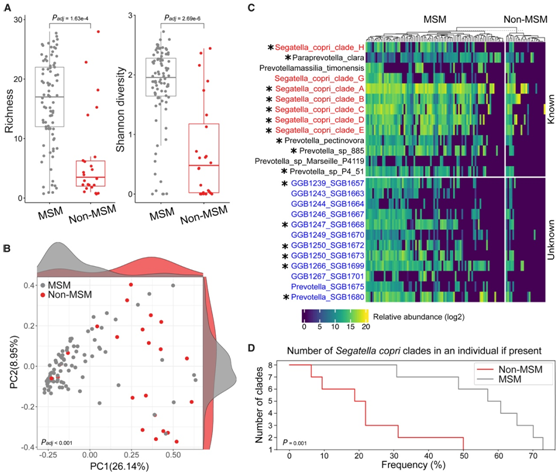
图2. MSM 和非 MSM 中的普雷沃氏菌群落结构。
(A) MSM 和非 MSM 中普雷沃氏菌科物种成员的丰富度和香农多样性。 (B) PCoA 基于MSM 和非 MSM 的普雷沃氏菌成员丰度。(C) 所有受试者中普雷沃氏菌科成员丰度。(D) 具有多个S. copri复合分支的个体的百分比。
03
MSM 肠道微生物群类似于Non-Wes 微生物群中存在的微生物多样性
为了进一步比较全球背景下的 MSM 微生物群结构,他们收集了来自 936 名健康成年个体的公开宏基因组数据集。在MSM中,Bacteroidaceae 和Bacteroides的丰度显著低于西方化(Wes)和中国城市人群,但显著高于Non-Wes样本(图3A)。相比之下,MSM的普雷沃菌科(Prevotellaceae)和Segatella/Prevotella丰度显著高于Wes受试者和中国城市受试者,但与非Wes受试者没有差异(图3A)。他们进一步将Prevotellaceae的比较扩展到整个微生物群落,结果表明,总体MSM微生物群落的特征与Wes和non-Wes样品不同(图3B)。在主坐标分析中,只有少数样本在Non-Wes样本和Wes样本之间分散,大部分样本与Non-Wes样本聚类,而与Wes样本聚类的样本数量较少(图3B)。这表明,与Non-Wes受试者相比,大多数MSM微生物组具有显著的相似性,特别是在Prevotellaceae/Segatella/Prevotella积累和Bacteroidaceae/Bacteroides消耗方面。结果表明,在各种群排名前50位物种中,MSM与Non-Wes个体共有33个物种,而MSM与Wes个体共有15个物种,与中国城市个体共有15个物种(图3C)。值得注意的是,在这项研究中,93名MSM中有92名有欧洲血统,并且都长期居住在德国(典型的Wes国家)。然而,还有其他微生物特征可能区分MSM与Wes和non-Wes个体。其中,相对于Wes和non-Wes样品,MSM样品中分别有216种和187种富集。除了多种S. copri复合物种和普雷沃菌属(Prevotella pectinovora)成员外,相对于Wes和非Wes个体,MSM中富集的微生物包括Megasphaera elsdenii、Megasphaera hexanoica和Paraprevotella clara(图3D)。
有趣的是,这些物种在粪便微生物群移植(FMT)过程中发生优先转移,这表明它们是可移植的属。有趣的是,一种名为芽囊原虫亚型1 (Blastocystis subtype 1)的真核寄生虫在Non-Wes群体中广泛分布,但在MSM人群中富集(图3D)。WES人群中富集的物种包括Akkermansia muciniphila、Streptococcus infantis和Erysipelatoclostridium ramosum(图3D)。非Wes人群中富集的物种包括Klebsiella pneumoniae、Veillonella parvula、Veillonella tobetsuensis和 Haemophilus parainfluenzae(图3D)。
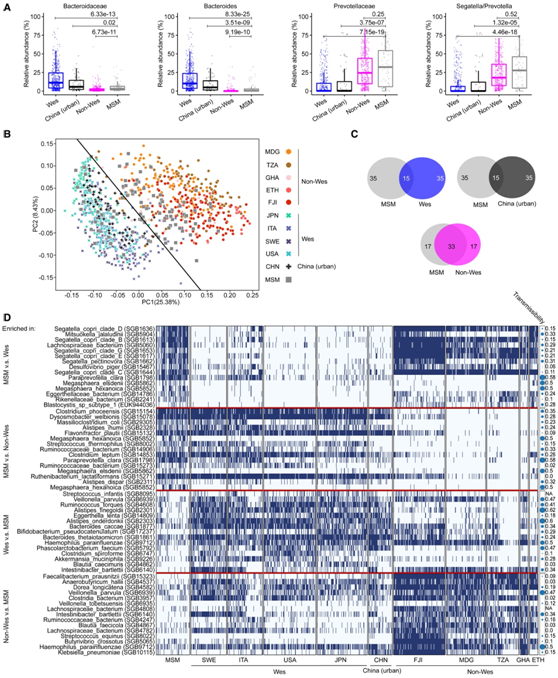
图3. 整体范围内 MSM 肠道微生物群落。
(A) MSM、西方化、非西方化和中国城市样本之间拟杆菌科和普雷沃氏菌科以及拟杆菌属和Segatella / Prevotella 属的丰度差异。(B) PCoA分析。 (C) 维恩图,分别显示 MSM 与西方化、非西方化和中国城市个体之间共有的和不同的物种数量。(D) 差异富集物种的存在-不存在的热图(蓝色,存在;白色,不存在)。
04
性行为与 MSM 肠道微生物组向非西方化变化相关
性行为会增加微生物感染的传播和发病率,例如艾滋病毒、病毒性肝炎和梅毒。然而,性行为对肠道微生物组的影响仍染是未知的。因此,他们在本项研究的队列子集中探索了这种联系,调查了性传播感染(STI)和性行为,包括接受性肛交(RAI)、性伴侣数量(过去 12 个月)、口交性别和安全套的使用。
由于α/β多样性和丰富度等汇总指标可能无法综合反映与性行为相关的微生物特征,因此接下来采用基于随机森林的机器学习方法来评估微生物群衍生信息对性行为提供的预测能力。在本项研究的案例中,目的是估计仅根据肠道微生物组分类组成将样本分类为相应的性行为类别(例如,拥有 >3 或 0-3 个伴侣)的准确度。值得注意的是,在所有性行为中都观察到了基于微生物组的预测能力(图4A),并且它在识别拥有 >3 个或 0-3 个伴侣的个体方面达到了最高性能(接受者操作特征曲线下的面积为 0.70)(图 4A-B)。
接下来,他们试图使用线性判别分析效应大小(LEfSe)来识别与性行为相关的微生物生物标志物。与风险降低行为(例如,在 RAI 期间始终使用安全套)相比,通常观察到与风险增加行为(例如,在 RAI 期间不使用安全套)相关的微生物生物标志物数量较多(图4D)。这表明,与降低风险的活动相比,增加风险的活动引入了更多区分肠道微生物组结构的微生物特征。拥有>3个性伴侣与多个Segatella/Prevotella成员共存有关,包括S. copri分支B和C以及P. pectinovora,这些成员在拥有>3个性伴侣的受试者中富集(图4C-D)。值得注意的是,大多数已确定的特定性行为的生物标志物仅与特定行为相关,然而,他们注意到一些生物标志物分别在风险增加和风险降低类别中对多种行为都是常见的(图4D)。例如,uSGB2230 (Rikenellaceae spp)是肛交接受者、口交从业者、性传播感染携带者(过去24个月)和RAI期间未使用安全套的人的重叠生物标志物(图4D)。相比之下,kSGB5075(Lachnospira pectinoschiza)与RAI期间一直使用避孕套并且(在过去12个月内)有0-3个性伴侣的人相互相关(图4D-E)。
接下来,他们分析了与风险增加相关的生物标志物是否比风险降低相关的生物标志物总体上更具传染性或更可能植入受体肠道,没有观察到水平传播率或传播相关特性(包括孢子形成、厌氧、运动性或革兰氏阴性)的显著差异。与风险降低行为相关的物种似乎显示出比风险增加行为相关的物种更高的定植率(图4F)。
最后,他们评估了在Wes社会中生活和长大的MSM的性行为与肠道菌群西方化或非西方化之间的关系。根据物种水平相对丰度的Bray-Curtis距离计算的主坐标,量化了MSM与Wes和Non-Wes种群的相似性(图3B)。有趣的是,风险增加的行为,如口交,有3个以上的性伴侣,以及在RAI期间不使用避孕套,与Non-Wes受试者的微生物组组成明显更接近(图4G)。这表明,特定的性行为与MSM中明显不同的微生物群有关,而这些微生物群与Non-Wes个体的肠道微生物组成具有相当大的共性。
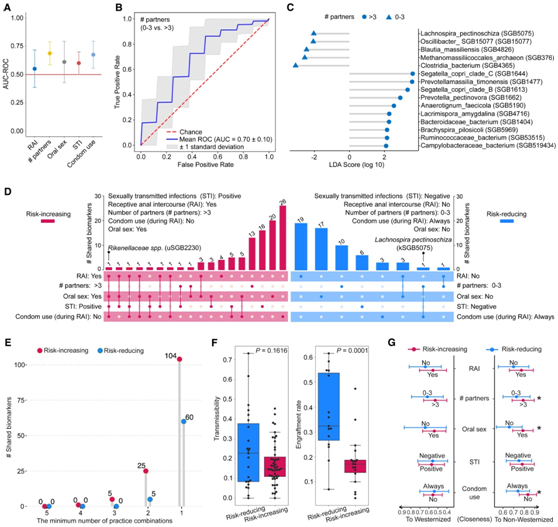
图4. MSM 肠道微生物组改变与性行为之间的关联。
(A) 通过机器学习预测评估的整个微生物组组成与性行为之间的关联分析。(B) 学习模型的 ROC(接收者操作特征)曲线,基于使用基于随机森林的方法。(C)使用LEfSe鉴定出的物种分类生物标志物与具有>3(和0-3)性伴侣的行为相关联。(D) UpSet 图显示使用 LEfSe 识别的分类生物标志物的重叠,将性行为分类为风险增加和减少。(E) 风险增加和减少类别中最少数量的实践组合所共有的分类生物标志物的数量。(F) 箱线图显示与风险增加和减少实践相关的分类生物标志物的传播率(左)和植入率(右)。 (G) 肠道微生物群与西方化和非西方化人群在性行为和性传播感染方面的密切程度。
05
MSM 肠道微生物组中未描述的微生物种类的富集
基于常用的质量标准,他们为代表765个物种的共计6065个推定基因组恢复了中质量和高质量基因组(图5A)。183个基因组(=株)属于72个先前未描述的物种,因为它们缺乏GTDB-Tk数据库的特征种水平分类分配。这些先前未描述的物种分布在本研究中重建的所有基因组所代表的几乎所有门中,除了Cyanobacteria、Elusimicrobiota、Methanobacteriota和 Myxococcota(图5A)。他们的菌株基因组在MSM样本中比在非MSM样本中更为普遍,86%的MSM具有至少一种先前未描述的菌株基因组,而在非MSM中这一比例为55%(图5A)。
为了估计 MSM 和非 MSM 样本中先前未描述的物种的菌株的读数丰度,他们试图将每个宏基因组样本的读数与 183 个先前未描述的菌株序列进行比对。MSM 样本具有的先前未描述的菌株特异性读数的比例要大得多(图 5 B)。76%的菌株基因组似乎比非MSM样本具有更多的读数(图5C)。这些结果揭示了MSM肠道微生物群比非MSM肠道微生物群含有更多尚未表征的细菌物种。
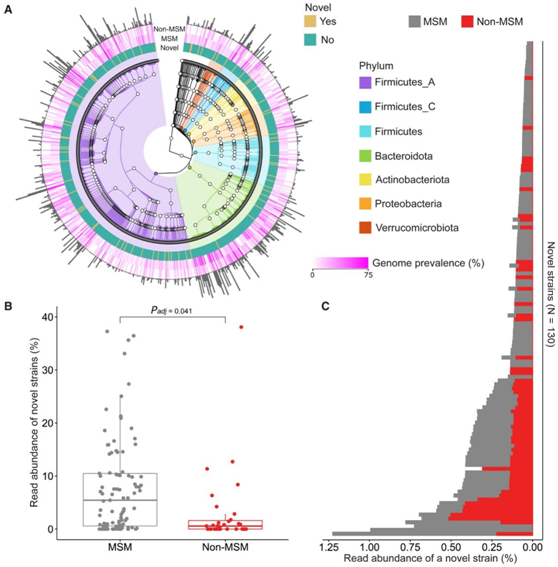
图5. MSM 和非 MSM 肠道微生物组中先前未描述的微生物种类的定量。
(A) 由从头宏基因组组装重建的 6,065 个假定基因组代表的 765 个物种的 GTDB-Tk 分类结构。(B) MSM 和非 MSM 宏基因组样本中先前未描述的菌株的丰度分布。 (C) 按菌株分层的读数丰度,平均分别超过 93 个 MSM 和 31 个非 MSM。
+ + + + + + + + + + +
结 论
本项研究通过宏基因组学进行物种水平的微生物群分析,揭示了许多西方起源的 MSM 的肠道微生物群与非西方化人群的肠道微生物群落相似。具体来说,MSM 肠道微生物组通常由普雷沃氏菌成员占主导地位,包括来自Segatella copri复合体的物种和未知普雷沃氏菌成员的共同定殖。机器学习还可以识别与 MSM 性活动相关的微生物特征。总之,这项研究显示了 MSM 中性活动与肠道微生物组改变的关联,这可能对基于人群的微生物组研究产生重大影响。
+ + + + +



