

 English
English文献解读|Nature(50.5):儿童 T 系急性淋巴细胞白血病的基因组分析
✦ +
+
论文ID
原名:The genomic basis of childhood T-lineage acute lymphoblastic leukaemia
译名:儿童 T 系急性淋巴细胞白血病的基因组分析
期刊:Nature
影响因子:50.5
发表时间:2024.08.14
DOI号:10.1038/s41586-024-07807-0
背 景
T 系急性淋巴细胞白血病 (T-ALL) 是一种高风险肿瘤,其基因组尚未得到全面的表征,部分原因是非编码基因组改变的频率很高,这导致致癌基因失调。
实验设计
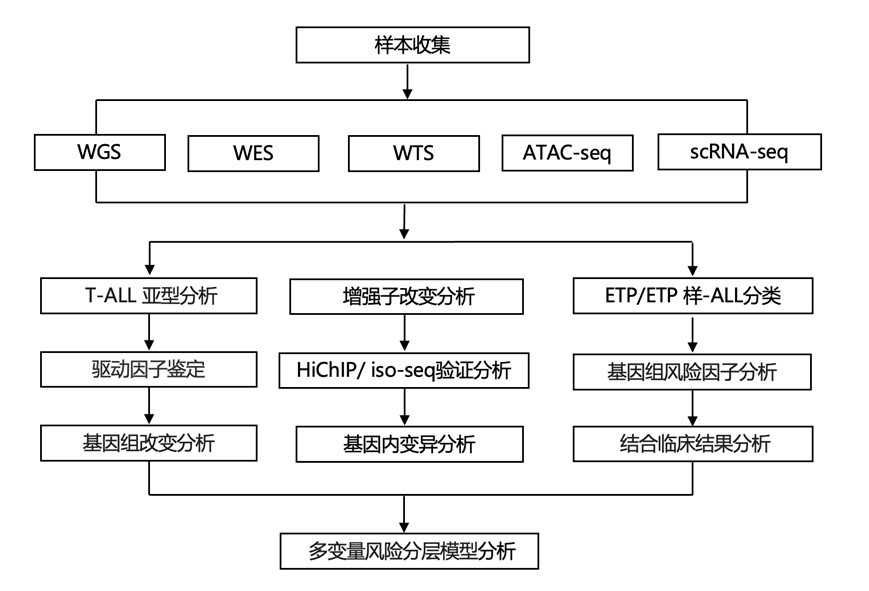
结 果
01

T-ALL的基因亚型分析
研究团队对儿童肿瘤队列(COG) AALL0434 试验中接受治疗的 1300 多名 T-ALL 患者的肿瘤和生殖系样本进行了全基因组测序 (WGS)、全外显子组测序 (WES) 、全转录组测序 (WTS)、单细胞转录组分析(scRNA-seq)和染色质转座酶可及性测序分析 (ATAC-seq) 。他们使用了均匀流形近似和投影 (UMAP) 技术和 Leiden 算法在二维中投影和聚类 WTS 基因表达数据(图1a,图S1)。鉴定了15 种具有不同驱动因子和致癌基因表达模式的 T-ALL 亚型(图S1b-e),包括 TAL1、TAL2、LMO1、LMO2 或 LYL1 转录调节因子失调或 TLX1、TLX3、NKX2-1 或 HOXA9 失调的亚型。在 95.1%(1309 例中的 1245 例)的病例中发现了假设的驱动因子,其中 59%(1309 例中的 777 例)位于基因组的非编码区域(图1b-c)。在4.9%的病例中未发现定义亚型的驱动因子,其中39%的肿瘤纯度低,聚集在髓系特征富集的组[肿瘤微环境(TME)富集组](图S1f)。
结合scRNA-seq和ATAC-seq数据,他们发现15 种 T-ALL 亚型涵盖了正常 T 细胞成熟的连续阶段(图1d,图S1g),包括从未成熟的造血干细胞 (HSC) 或造血干细胞和祖细胞 (HSPC) 到常见的淋巴祖细胞 (CLP)、淋巴髓样引发祖细胞 (LMPP) 或 BCL11B 和早期 T 细胞前体 (ETP)样亚型细胞。pro/pre-T 细胞与 KMT2A、TLX3 和 MLLT10 亚型相似。循环双阳性 (DP) 细胞类似于 HOXA9 T 细胞受体 (TCR)、TLX1、NKX2-1 和 TAL1 DP 样亚型。表达 TCRA的单阳性/成熟 αβ 细胞类似于 TAL1 αβ 样亚型。最后,γδ/效应 T 细胞类似于 LMO2 γδ 样亚型,后者也与 TCRγδ 重排有关(图S1h)。他们还观察到髓系特征的富集,包括 SPI1 亚型的树突状细胞 (DC)、NKX2-5 的粒细胞/单核细胞祖细胞 (GMP) 和 STAG2/LMO2 亚型的巨核细胞/红细胞前体 (MEP) 细胞。
他们在之前识别的亚型中发现了其他基因组改变(图2)。例如,TLX3亚型中TLX3的失调通常是由于劫持(hijacking)了BCL11B (ThymoD)增强子,但他们发现了多个发生重排和TLX3失调的额外增强子,包括TCRβ位点、CDK6、notch1驱动的MYC增强子(N-Me)和SATB1增强子。
根据基因组和临床特征,他们将几种亚型划分为亚组。TAL1、TAL2、LMO1、LMO2 或 LYL1 核心转录回路失调的病例可分为两种亚型,先前称为 TAL1-RA 和 TAL1-RB 。考虑到本研究的肿瘤和正常祖细胞基因组数据,他们将这些亚型称为 TAL1 αβ 样和 TAL1 DP 样亚型。TAL1 DP 样亚型表现出更高的RAG1、RAG2、CD4和CD8表达,而 TAL1 αβ 样亚型的特点是 TCRα 常数 (TRAC) 基因表达更高和 TCRα/β 重排(图2,图S1h)。这些亚型中已知的驱动因子是STIL::TAL1,TAL1、LMO1、LMO2和LYL1重排为 TCRδ/β 增强子,以及产生TAL1新增强子的非编码序列突变。他们确定了多种其他致癌基因失调机制,包括拷贝数变异(CNV) 和单核苷酸变异 (SNV)和插入/缺失突变(indel),这些突变会产生TAL1、LMO2和LYL1的新增强子,以及导致增强子劫持(enhancer hijacking)介导的TAL1和LMO2失调的基因间倒位和缺失。
他们定义了另外两种亚型。ETP 样亚型富含 ETP 免疫表型病例,并具有多种基因组驱动变异。LMO2 γδ样亚型具有多种变异,包括BCL11B增强子劫持、FOXP1重排、 MYC重排为 TCRδ 以及增强子SNV /indel 激活LMO2(图S1h -j)。
他们观察到亚型与临床特征之间的关联。STAG2/LMO2、NKX2-5 和 SPI1 T-ALL 组患者诊断时年龄较小,而 BCL11B 和 HOXA9 TCR 亚型中老年患者的比例更高(图S1k-l)。不同亚型的性别分布不同,STAG2/LMO2、ETP-样、HOXA9 TCR、LMO2 γδ-样和 NKX2-5 亚型中女性患者比例较高,TAL1 亚型中男性患者比例较高(图S1m)。总的来说,这些研究结果表明 T-ALL 亚型主要由非编码驱动基因改变和细胞分化阶段来划分。
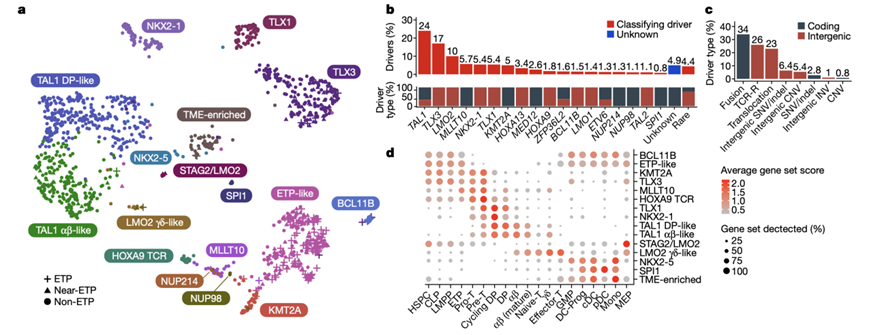
图1. 根据驱动因子和基因表达进行分类,定义了 15 种 T-ALL 亚型。
(a) UMAP 散点图描绘了 T-ALL 亚型和 ETP 状态。(b) “未知”病例中分类驱动因子改变的百分比和每个驱动因子的改变比例。(c) 驱动因子改变类型百分比。 (d) 正常 BM 和胸腺 scRNA 数据中亚型基因表达特征的基因集得分。
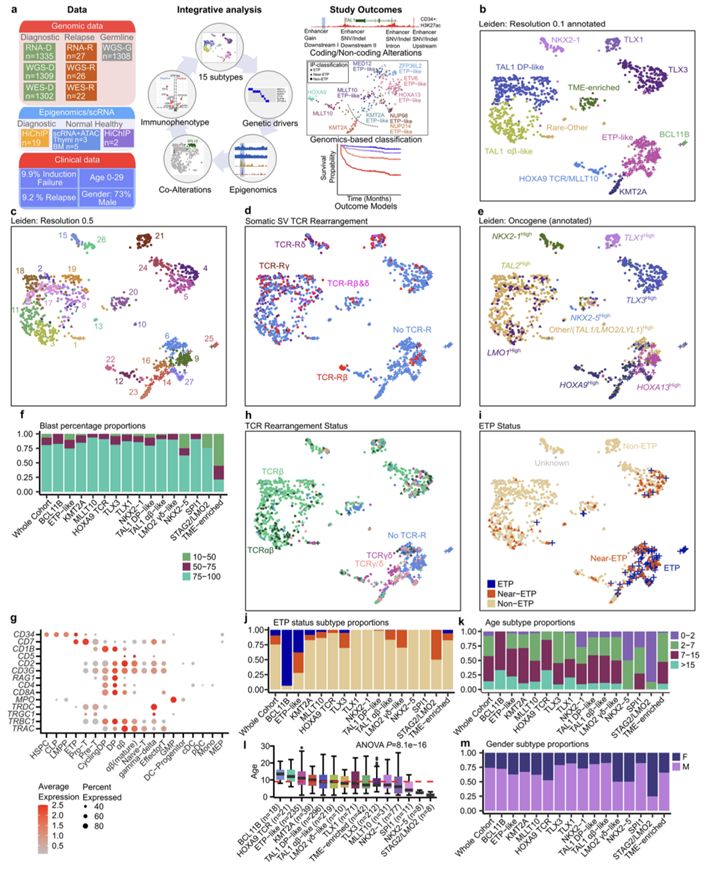
图S1. T-ALL队列组成和亚型。
(a) 队列组成、分析框架和研究结果的示意图。(b) UMAP散点图显示0.1分辨率的Leiden算法聚类。(c) Leiden算法以0.5分辨率聚类,进一步描绘不同的组。(d) UMAP图显示了通过增强子导致癌基因激活的结构变异(SV)中涉及的TCR位点类型。(e) Leiden算法聚类显示癌基因表达模式。(f) 按亚型分类展示原始细胞百分比分布比例的柱状图。(g) 标记基因的点图。(h) UMAP图显示了癌细胞中克隆性TCR重排的鉴定。 (i) ETP状态的UMAP图。(j) 具有已知免疫表型的病例,按亚型分类的ETP状态比例条形图。(k) 条形图显示年龄分布比例,按亚型分类并按中位年龄排序。(l) 各亚型的年龄分布。(m) 柱状图描绘性别分布比例,按亚型分类。
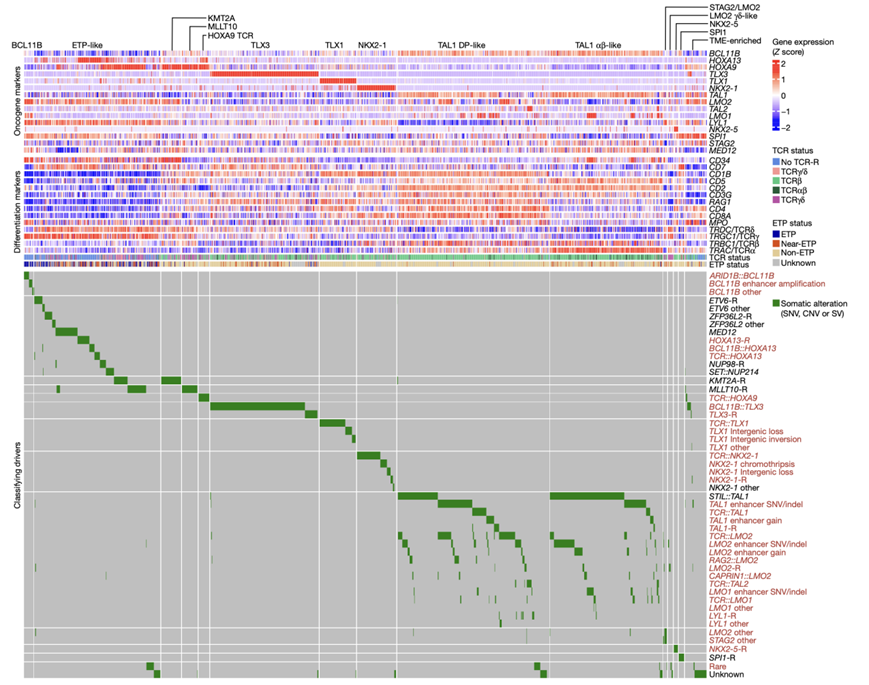
图2. 对驱动因子进行分类的肿瘤印迹。
02
癌基因激活增强子改变
他们在 70.5% 的病例中观察到增强子介导的致癌基因激活,在 43.9% 的病例中需要 WGS 才能检测到这些激活。这些激活包括易位、倒位、染色体碎裂和缺失,在 50.4% 的病例中观察到 12 个增强子与 61 个致癌基因并列,在 34.8% 的病例中序列改变导致 8 个基因产生新增强子(图3a)。他们对 19 个 T-ALL 样本(包含 6 种亚型的 19 种不同改变)使用了 ATAC-seq 和组蛋白 3 赖氨酸乙酰化 (H3K27ac) HiChIP 技术,以检测染色质的可及性以及与致癌基因的相互作用和假定的增强子。通过 HiChIP 验证了50 个基因,其中 23 个发生了显著突变、劫持的 TCR 增强子以及失调HOXA13、HOXA9和LMO3 的事件。
他们检测到TAL1下游 28 kb 处反复出现的TAL1增强子增益(中位大小 133 bp), TAL1下游 20 kb 处增强子SNV / indel,TAL1第一个内含子中的 SNV/indel以及TAL1上游的增强子插入缺失,它们都是 CD34 +细胞而非 DP 细胞中的活性增强子(图3b)。他们使用 ATAC-seq、HiChIP 和异构体测序(iso-seq)验证了TAL1增强子增益(图3c)。
几个增强子劫持事件与发育阶段有关。TAL1基因间易位至CD1基因座导致CD1E增强子发生劫持,这种增强子在正常 DP 细胞和胸腺 CD34+CD1a+细胞中活性较高,但在 BM CD34 +细胞中活性较低。同样,在 TAL1 DP 样亚型中观察到LMO2和RAG2之间的基因间缺失,驱动LMO2失调的RAG2增强子在正常 DP 细胞中活性较高,而在 CD34 +细胞中活性较低(图3d)。相反,ETP 样亚型中HOXA13失调的病例表现出位于CASC15基因座内的SOX4增强子的劫持,这些增强子在正常 BM 和胸腺 CD34+ 细胞中的活性高于 DP 细胞,并且经常劫持在 γ/δ 或 CD34+细胞中的MIR181A1增强子(图3e)。
他们在 50 个基因中观察到了非编码和基因内事件,他们已证明或预测这些基因具有多种基因激活和扰动机制。例如,NOTCH1经常含有激活 NOTCH1 信号传导的编码序列突变。他们还在 1.6% 的病例中鉴定了内含子 SNV,这些 SNV 在内含子 28 中产生非规范剪接受体,导致外显子 28 编码序列的近端延伸(图3f-g)。与常见的编码突变相比,内含子SNV驱动了NOTCH1活性的增加(图3h)。
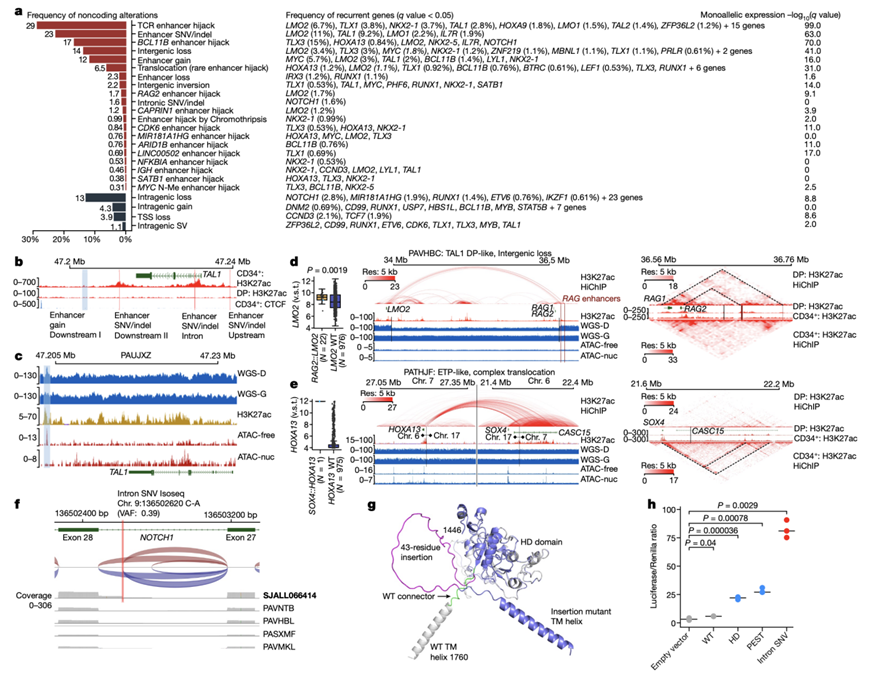
图3. T-ALL 中致癌基因和增强子驱动改变的多种机制。
(a) 左图为非编码基因间和基因内变异。右图为反复出现的靶基因。(b) TAL1增强子变异,CD34 + HSPC 和 DP T 细胞 H3K27ac 和 CTCF 覆盖率。(c) 对于显示出TAL1增强子增益的样本,诊断(D)和生殖系(G)全基因组测序(WGS)、H3K27ac、无核小体和ATAC-seq核小体切割片段(ATAC-free、ATAC-nuc)的覆盖情况。(d) 在因基因间区丢失导致RAG2::LMO2的PAVHBC中,H3K27ac HiChIP、全基因组测序(WGS)和ATAC-seq的分析。(e) 与(d)相同,但针对染色体7与染色体6及染色体7与染色体17易位导致的SOX4::HOXA13增强子劫持(enhancer hijacking)的情况。(f) 针对具有突变的患者样本和 WT 对照样本的NOTCH1外显子 28-29 内含子 SNV 的剪接连接读取和覆盖率。(g) AlphaFold2模型显示了WT和NOTCH1内含子SNV突变体之间的结构差异。(h) NOTCH1内含子SNV、HD和PEST结构域突变和对照的相对荧光素酶活性。
03
ETP 样亚型的特征
他们在 ETP 样病例中观察到的MED12改变遍及MED12的编码区,这表明功能丧失。为了验证该结果,他们使用基因组编辑在 LOUCY(SET::NUP214)细胞系中使MED12失活,该细胞系与 ETP-ALL 具有免疫表型相似性。他们观察到该模型中组蛋白去乙酰化酶途径基因表达的上调。将这些数据与MED12改变的 ETP 样病例的基因表达谱相结合,结果显示 T 细胞分化标志物CD5和CD28的表达共同降低,干细胞标志物LMO2和HHEX的表达增加(图4a-b)。这些结果表明 MED12 功能的丧失直接导致 ETP 类 ALL 的不成熟特征。
整合基因组分析还揭示了在 ETP 样 ALL 中驱动特定HOXA基因差异失调的机制。具体而言,驱动HOXA9而非HOXA13失调的增强子劫持改变(如TCRB::HOXA9)表明重排断点始终位于两个 CTCF 峰之间,这两个峰划定了CD34+ HSPC 中HOXA9和HOXA13基因座之间的拓扑关联域 (TAD) 边界(图4c)。相比之下,所有导致HOXA13失调的重排的断点都局限于 HOXA13 TAD,从而限制了HOXA9的激活。
虽然28% 的 ETP 样亚型病例不符合 ETP样 ETP-ALL 的免疫表型标准,但它们表现出相似的免疫表型趋势(T 细胞标志物和髓系和干细胞标志物表达较低),包括不存在 TCR 重排、相似成熟阶段、基因组驱动因子和预后(图4d-i)。
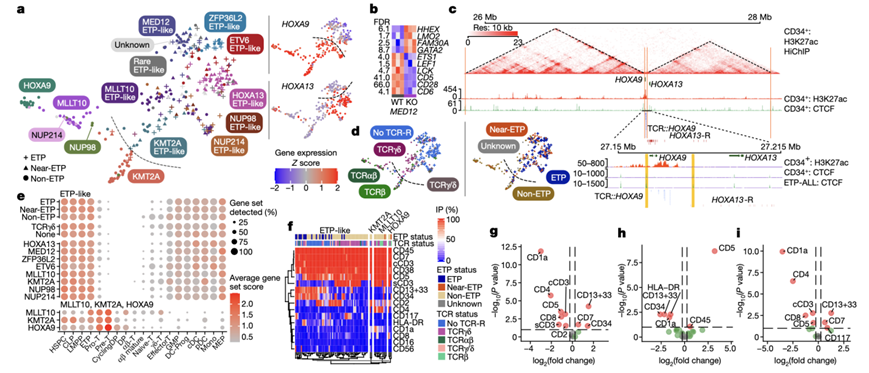
图4. ETP/ETP 样 ALL 的基因组分类。
(a) 左,ETP 样基因亚组以及 HOXA9、MLLT10 和 KMT2A 组分布的 UMAP。右,HOXA9和HOXA13表达Z 分数。(b) 差异表达基因的热图。(c) 正常CD34+造血干细胞(HSPC)中,H3K27ac HiChIP原始相互作用数据以及H3K27ac和CTCF的覆盖情况,显示出HOXA9和HOXA13具有不同的拓扑关联域(TAD)。 (d) ETP状态和TCR-R状态在类ETP样本中的分布的UMAP。(e) 基因集富集的点图。(f) ETP 样病例以及 KMT2A、MLLT10 和 HOXA9 亚型的免疫表型 (IP) 细胞表面标志物表达的热图。(g) 火山图表示 ETP 样病例与其余样本之间的差异流式细胞标志物。(h-i) 差异标记。
04
基因组风险因子的结果分析
他们研究了与临床结果相关的基因组特征。残留疾病(RD)和微小残留疾病(MRD)≥ 0.01%)的阳性率在 ETP 样和 LMO2 γδ 样亚型中尤为常见(图5a)。他们研究了亚型、遗传驱动因子、共同病变、广泛的拷贝数变异(CNV)和改变的通路与 RD 之间的关联。ETP 样驱动因子和共同病变(SH2B3、ETV6、NRAS和WT1)与较高的 RD 风险相关。相比之下,TAL1 亚型相关特征(LEF1、USP7、PI3K和CCND3)与较低的 RD 风险相关。 JAK–STAT 和 RAS 信号通路改变与较高的 RD 风险相关,而 NOTCH、核糖体和 PI3K 通路与较低的 RD 风险相关。
在无事件生存期 (EFS)、无病生存期 (DFS) 和总生存期 (OS) 分析中,SPI1 和 LMO2 γδ 样亚型预后较差。NKX2-5 亚型和 ETP 样 KMT2A、MLLT10 和 HOXA13 基因亚型也有不良预后(图5b)。非 ETP 样 KMT2A 亚型的 MRD 较高,但预后良好,与 ETP 样亚型中的 KMT2A 病例不同。他们观察到单个基因的特定类型的遗传变异会调节结果(图5c),大多数NOTCH1变异具有良好的预后,但内含子 SNV 和基因内丢失与较差的 OS 和 EFS 相关(图5c)。TCR::MYC重排的病例 DFS 较差,而MYC增强子增加的病例 DFS 较好。与其他 PI3K 通路改变相比, PTEN缺失的病例结果明显更差。在 TAL1 亚型中,TCR:: LYL1和LMO2基因间丢失导致RAG/CAPRIN1增强子劫持与较差的结果相关,这与 6q 缺失和RPL10突变形成对比(图5c)。
第二种恶性肿瘤并不常见,但以 SPI1 亚型为主,其中 11 名患者中有 4 名在诊断后一年内发展为致死性组织细胞增生症或髓样肉瘤(图5d)。WGS 和 WTS数据证实,在诊断性 T-ALL 和 LCH 中,起始克隆均含有STMN1::SPI1、CDKN2A缺失和KRAS突变(图5e)。T-ALL样本还表达DC基因(HLA-D、CD1a、CD38、CD45、CD7、CD5和sCD3), SPI1 T-ALL样本显示富集了正常DC特征(图5f-g)。这些发现表明,SPI1 融合驱动的恶性肿瘤出现在具有 T 细胞和 DC 潜力的祖细胞中,并经常转变为高风险的 DC 相关的第二恶性肿瘤。
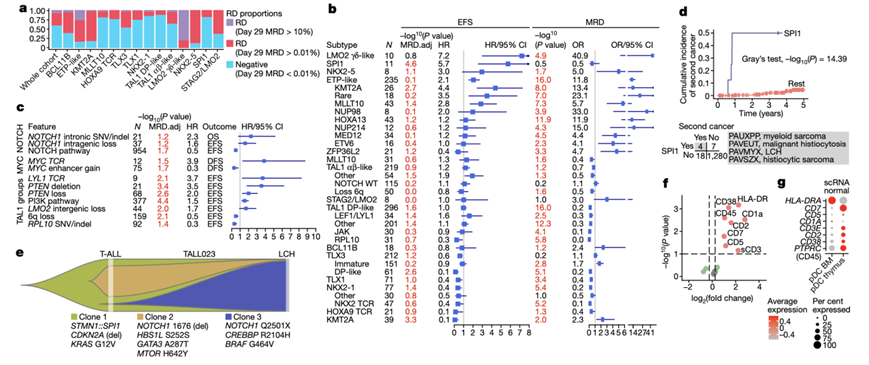
图5. T-ALL 基因组风险因子和临床结果。
(a) 每种亚型的残留疾病 (RD) 类别比例。(b) 森林图以箱线图 (HR) 的形式显示风险比。(c) 选定变体的森林图。(d) SPI1 融合和第二种癌症的累积发生率图。(e) 病例 TALL023 中 T-ALL 向 LCH 的克隆进化,每个克隆显示 T-ALL 驱动改变。(f) 火山图描绘了 SPI1 亚型病例与其他样品之间的差异标记物。 (g) 点图显示BM 和胸腺 DC 中来自(f)的免疫表型标记物。
05
多变量风险分层模型
他们探索了多种多变量建模方法的实用性,这些方法结合了临床变量、治疗反应和基因组特征,以预测结果并对患者进行风险分层。当使用数值 MRD、临床变量(性别、诊断时白细胞计数 >2 × 105细胞/μl 和中枢神经系统状态)和亚型/变异水平基因组特征(pCox 模型)和遗传亚型(RSF 模型)拟合每个模型时,随机生存森林 (RSF) 和惩罚 Cox 回归 (pCox)模型具有最高的准确率。
pCox 模型结合 3 种临床特征、5 种亚型和 18 种基因组改变,将患者分成四个大小相等的组,5 年 EFS 范围为 65% 至 97%(图6a-b)。SPI1 亚型、ETP 样亚型、PTEN缺失或丢失、PIK3CD SNV/插入缺失和LMO2基因间丢失与较差的结果相关,而 KMT2A 亚型、6q 丢失、RPL10和NOTCH1 SNV/ indel在单变量和 pCox 模型中均与有利的结果相关。这些结果凸显了它们作为独立预后生物标志物的价值(图5b-c和6a)。
他们将RSF 模型简化为四节点生存树 (ST) 模型,该模型通过有针对性地选择特征可用于临床实施。ST 模型能够将患者风险分层为 8 组,5 年 EFS 范围从 45% 到 98%(图6c)。包括 ETP 样驱动基因 KMT2A、MLLT10、NUP98 和稀有驱动基因以及 SPI1、LMO2 γδ 样和 NKX2-5 亚组在内的几种特征无论 MRD 反应如何,预后均较差(5 年 EFS < 60%)。这些患者应考虑进行 HSC 移植或新的免疫疗法,因为即使进行了多药强化化疗,预后仍然较差。相比之下,如果第 29 天 MRD 小于 0.1%,其他几个特征(包括 ETP 样ZFP36L2变异、TLX3 DP 样、TAL1 DP 样 RPL10、NKX2-1、TLX1、KMT2A 和 HOXA9 TCR)具有较好的预后(5 年 EFS > 98%),这群庞大的患者群体可能会从化疗强度的降低中受益。
事实证明,pCox 模型和 ST 均能有效准确预测 ETP 样组和按免疫表型分类的组中个体的结果(图6c)。相比之下,ETP 样亚型的免疫表型不具有预后意义,而 BCL11B 亚型即使免疫表型为 ETP 也具有良好的结果(图5b)。这些发现强调了使用基于基因组的多变量预后分类的必要性。
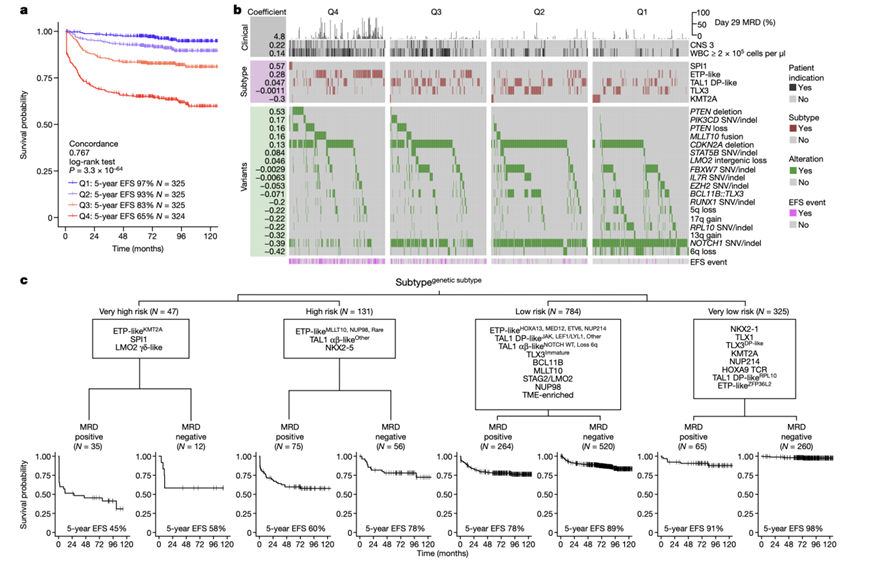
图6. 多变量结果模型。
(a)生存图。(b) 基因组或临床特征。(c) 拟合的生存树 (ST) 模型。
+ + + + + + + + + + +
结 论
本项研究对1300多名接受统一治疗的 T-ALL 儿童的肿瘤和缓解样本进行多组学分析,确定了 15 种具有不同基因组驱动因子、基因表达模式、发育状态和结果的亚型。染色质拓扑分析揭示了增强子失调的多种机制,这些机制以亚型特异性的方式涉及增强子和基因,从而证明非编码基因组的广泛参与。多变量结果模型分析显示遗传亚型、驱动因子和伴随遗传变异独立地预测治疗失败和预后。这些发现为该疾病的分类、风险分层和机制理解提供了新的理论依据。
+ + + + +



