

 English
English文献解读|Nat Commun(16.6):综合多组学分析揭示 WEE1 通过 CDK1 超级激活与高热协同致死靶点
✦ +
+
论文ID
原名:Comprehensive multi-omics analysis reveals WEE1 as a synergistic lethal target with hyperthermia through CDK1 super-activation
译名:综合多组学分析揭示 WEE1 通过 CDK1 超级激活与高热协同致死靶点
期刊:Nature Communications
影响因子:16.6
发表时间:2024.03.07
DOI号:10.1038/s41467-024-46358-w
背 景
上皮性卵巢癌(EOC)通常出现在晚期,仍然是最致命的妇科癌症。造成EOC高死亡率/发病率的主要因素之一是初始诊断时已进入晚期,伴有广泛的腹膜转移 (PM),并且缺乏对晚期疾病的有效治疗。荷兰一项精心设计的随机试验 (OVIHIPEC 1) 显示,结合热疗(HT)和腹腔化疗的高温腹腔化疗(HIPEC),在接受最佳间隔减体积手术的EOC患者中,可显著改善总体生存率(OS),但HIPEC在卵巢癌中的作用仍然存在争议,且目前对高温引起的肿瘤细胞变化的了解有限。
实验设计
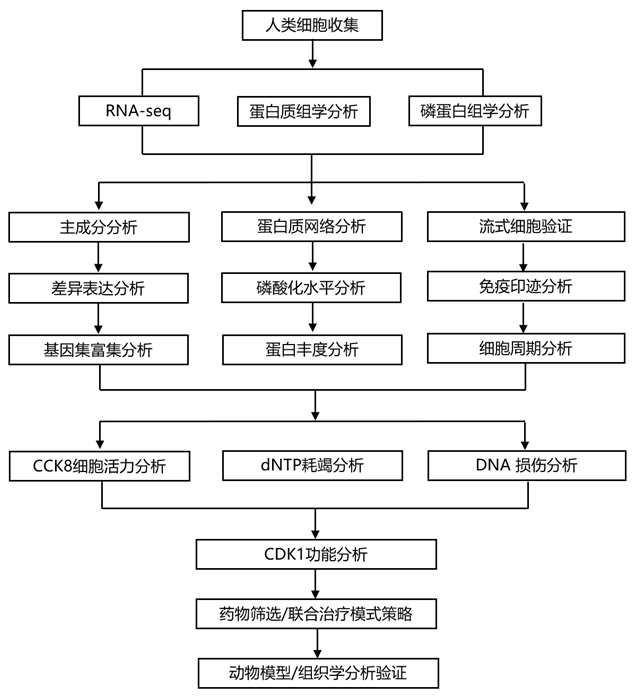
结 果
01

多组学整合分析揭示高温导致上皮性卵巢癌细胞中 CDK1 过度激活
研究团队在常温(37℃)和高温(42℃,90分钟)条件下,分析了卵巢癌中以TP53突变为特征的细胞系OVCAR8细胞的转录组、蛋白质组和磷酸化蛋白质组(图1a)。
他们定量了16954个转录本,6625个蛋白质和10725个磷酸化位点(图S1a)。转录组、蛋白质组和磷酸化蛋白质组数据的主成分分析 (PCA) 分别显示了重复之间的高度一致性(图S1b)。就个体而言,差异表达(DE)分析显示692个转录本(4.1%)、322个蛋白(4.9%)和2178个磷酸化位点(20.3%)对HT有响应(图S1c)。基因集富集分析(GSEA)显示,高温反应转录本聚集在与热反应或蛋白质折叠相关的通路上(图S1d)。在本项研究的蛋白质组学分析中也鉴定了6570个转录本(图S1e),表明两种分析发现的基因/蛋白质有显著的重叠。尽管RNA和蛋白质丰度的总体相关性很高,但通过直接比较相同条件下的差异蛋白表达和mRNA变化,他们发现HT诱导的转录组和蛋白质组变化之间的相关性较差(图S1f-g),这与广泛的转录后调控一致。总体而言,与mRNA水平相比,HT显著调节的蛋白数量要少得多,53个蛋白增加,269个蛋白减少(图S1f),只有14个差异表达的RNA和蛋白(10个上调,包括HSPA6、HSPA1B、DEDD2、IER5、DUSP1;4个下调,包括DKK1和CCN1)与转录反应重叠(图S1f-g)。值得注意的是,在丰度显著增加的少数蛋白中,绝大多数富集于细胞周期通路(E2F、G2M检查点和有丝分裂纺锤体)(图S1h-i)。
磷酸化变化发生迅速,而蛋白质变化需要更多的时间来发生,因此大多数受到显著调节的分子事件发生在磷酸化水平而不是蛋白质丰度水平(图S1c)。在HT处理的细胞中,共有2178个磷酸化肽,对应于1344个蛋白质,表明HT对蛋白质磷酸化有实质性的影响。重要的是,很少的磷酸化位点(2.0%)在蛋白水平上有任何变化,进一步表明磷酸化信号通路代表了HT这一时间过程中的主要细胞反应(图S1j)。
他们接下来基于已知的激酶和底物关系研究了激酶信号级联的差异调节及其对蛋白磷酸化的影响。预计在高温下最强烈激活的激酶包括几种细胞周期激酶(CDK1、AURKB和CDK2)(图1b)。预计下调的激酶包括CK2A1、GSK3B和MTOR(图1b)。CDK1的底物(TOP1pS394、TP53BP1pT1609、CDC25CpT48、USP14pT235等)在HT后的上调磷酸化蛋白中显著过表达。
综合分析显示,CDK1依赖性细胞周期检查点是HT后最受干扰的通路,包括其自身的抑制性Tyr15 (Y15)磷酸化显著降低(图1d)。他们证实了HT诱导CDK1对Thr14 (T14)和Y15的抑制性磷酸化的时间依赖性降低,而对OVCAR8、A2780和ID8细胞的总蛋白水平没有明显影响(图1e)。磷酸化-Thr320 PP1Cα (PP1Cα-pT320)可作为CDK1活性的指标,通过免疫印迹法(western blotting)评估,HT后这种修饰以时间依赖性的方式逐渐增加(图1e)。此外,磷酸化-Ser139 H2AX (γH2AX)大量增加,表明HT诱导的复制应激最终导致DNA损伤(图1e)。
CDK1控制关键的细胞周期事件,如DNA复制和G2/ M期转变。过度激活CDK1通过依赖CDK1的复制起始点异常激活诱导复制应激。因此,他们采用5-乙基-2 ' -脱氧尿苷(EdU)脉冲标记流式细胞追踪HT后细胞周期进程和DNA复制过程。与CDK1在G2/M细胞周期检查点上的关键作用一致,HT增加了G2/M细胞群(图1f)。此外,他们观察到高温应激状态下OVCAR8、A2780和ID8细胞积累了非复制的S期细胞(NS期,表现出DNA含量在2N和4N之间,但不含合成核苷EdU),这表明了高温后DNA复制应激发生(图1f)。然后,他们通过DNA纤维测定来描述单个复制叉的进展,DNA纤维测定是监测复制叉速度进展和起源的金标准测定。HT处理后OVCAR8细胞的复制叉速度显著降低(从平均0.6-0.8 kb/min降低到0.3-0.5 kb/min)(图1g),进一步证明HT诱导复制叉停滞。停滞的DNA复制分叉导致单链DNA (ssDNA)暴露。
为了进一步证实过度激活的CDK1是HT后周期扰动、复制应激和DNA损伤积累的原因,他们在HT处理的细胞中添加了CDK1抑制剂(CDK1i, RO-3306)。经HT处理后,RO-3306可恢复多种卵巢癌细胞系的细胞活力(图S1k)。此外,RO-3306还能消除HT诱导的G2/M细胞群积累,减轻非复制S期和复制应激(图1h,图S1l)。值得注意的是,HT诱导的DNA损伤(γH2AX)主要分布在G2/M期和非复制S期,与复制停滞和有丝分裂突变导致DNA损伤一致(图1h,图S1l)。此外,在非变性条件下,BrdU染色的OVCAR8细胞在HT后观察到大量ssDNA的积累(图1i)。值得注意的是,许多BrdU阳性细胞显示同时弥漫的泛核γH2AX染色,而不是点状灶(图1i),表明灾难性的复制应激和不可逆的细胞死亡。正如预期的那样,RO-3306显著阻断了γH2AX和ssDNA的诱导(图1i)。
为了确定其普遍性,他们在另外13种具有不同遗传背景的细胞系(8种卵巢癌、2种结肠癌和3种非肿瘤细胞)中检测了HT后的分子变化(图S1m)。在所有癌细胞中,磷酸化Tyr15 CDK1 (CDK1- pY15)下调,DNA损伤增加。相比之下,在非肿瘤细胞IOSE80(正常卵巢表面上皮细胞)、HUVEC(人脐静脉内皮细胞)和HaCaT(人角质细胞)中,CDK1-pY15和γH2AX在HT处理后未见明显变化(图S1m)。鉴于热休克转录因子1 (HSF1)介导细胞应激反应中不可或缺的热休克蛋白的转录,他们利用CRISPR-Cas9建立了HSF1敲除SKOV3细胞(图S1n)。这些研究结果表明,HT后的CDK1激活不依赖于HSF1(图S1o)。
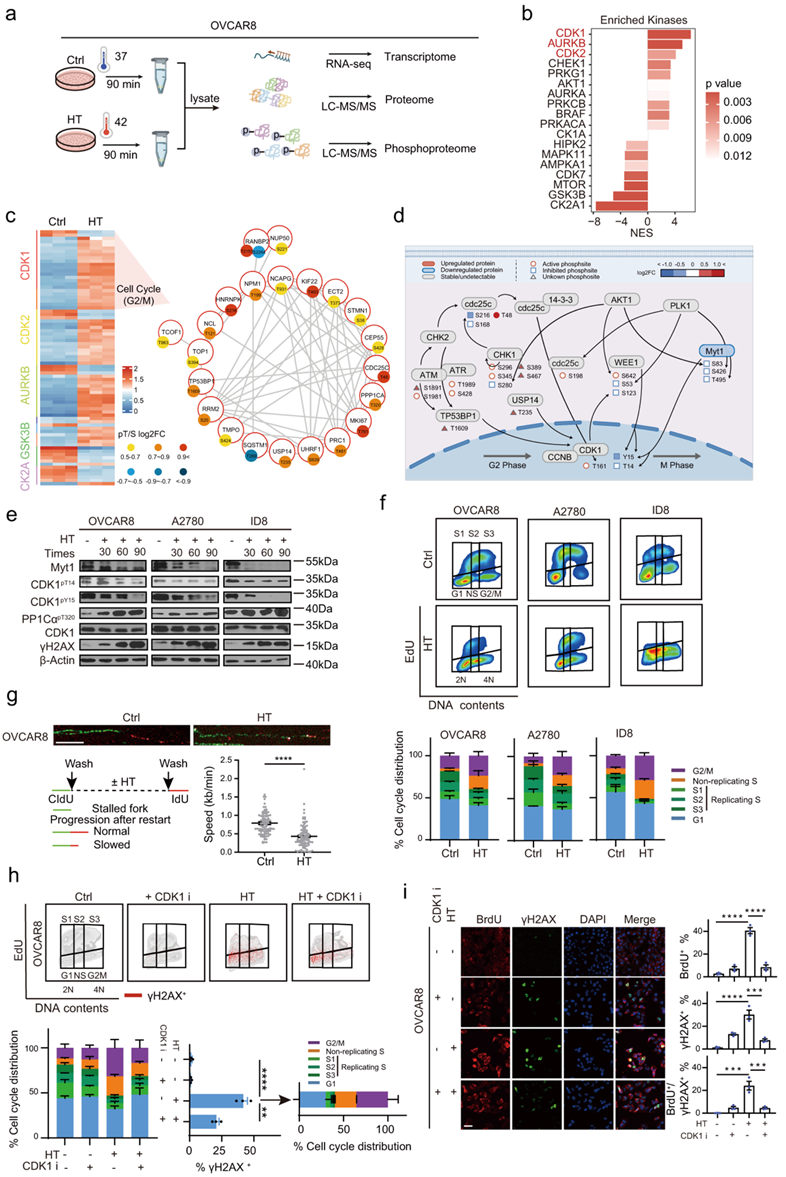
图1. 高温导致cdk1依赖性细胞周期紊乱、复制应激和DNA损伤。
(a) 转录组、蛋白质组和磷酸蛋白质组分析的实验方案。 (b) 柱状图显示了高温(42°C, HT)和常温(37°C, Ctrl)处理的OVCAR8细胞的不同磷酸位点,通过PTM-SEA进行分析。(c) 热图显示了与部分(b)富集的激酶相关的差异调节磷酸化位点(左)。CDK1差异底物的蛋白-蛋白相互作用网络,来自STRING数据库(右)。(d) HT诱导cdk1依赖性细胞周期相关蛋白和磷酸化水平的变化概述。 (e) 免疫印迹。(f) 高温暴露90min后OVCAR8、A2780和ID8细胞系细胞周期分布。(g) 对(f)部分处理的OVCAR8细胞进行DNA纤维分析,得到具有代表性的复制叉图像,比例尺为10 μm。 (h) 流式细胞分析。(i) 为了检测ssDNA,整个实验过程中,细胞在10 μM BrdU的培养基中培养;BrdU染色前DNA未变性。图中显示了BrdU、γH2AX和DAPI染色的OVCAR8细胞的代表性图像和定量。
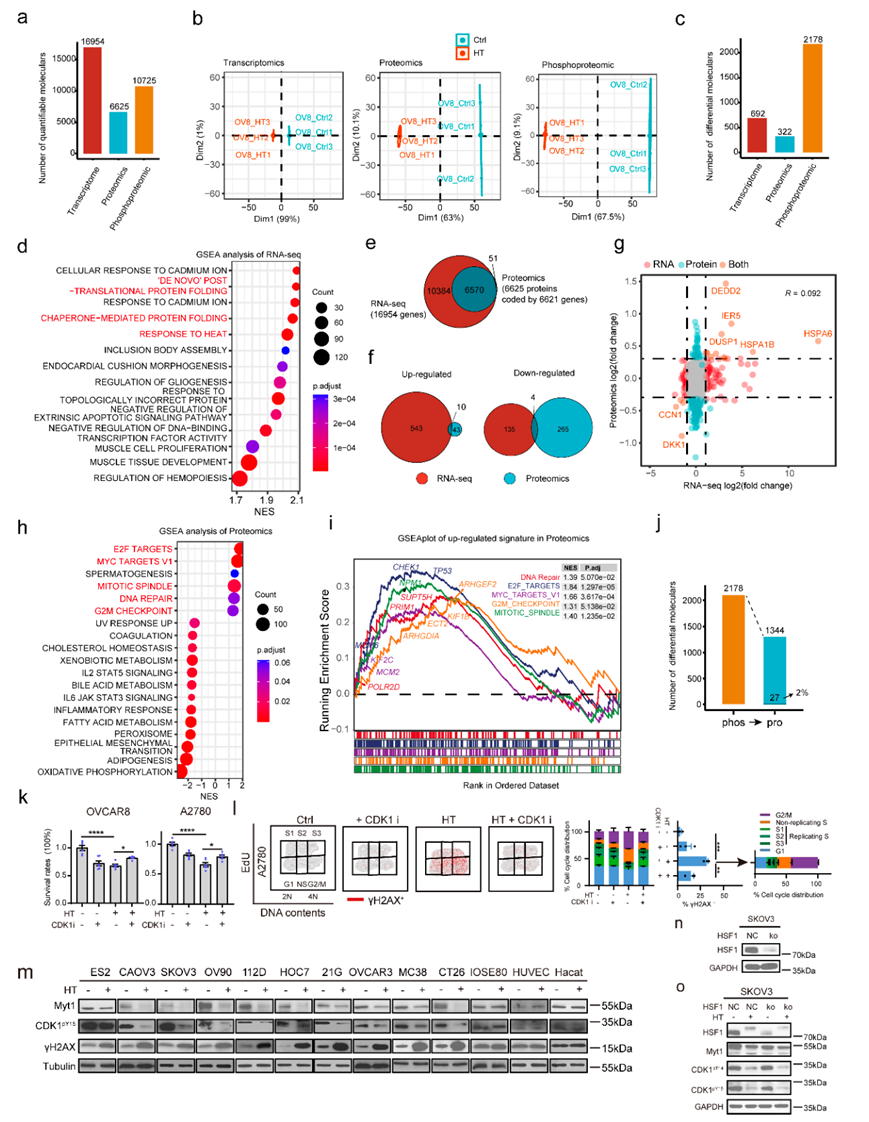
图S1.多组学分析。
(a)条形图显示转录组学、蛋白质组学和磷酸化蛋白质组学中定量的基因数量。(b) 主成分(PCA)分析。(c) HT后RNA、蛋白质和磷酸化水平的差异表达分析。(d) GSEA 分析。(e) 显示蛋白质组学中可定量蛋白和转录组中mRNA重叠的维恩图。(f) 维恩图显示了转录组学和蛋白质组学中HT后上调(左)或下调(右)的分子重叠。(g) 散点图显示蛋白质和RNA表达变化的重叠和相关性。(h) 采用蛋白质组学方法进行GSEA分析。(i) 蛋白质组学GSEA图显示HT后信号通路受到显著干扰。(j) 条形图显示了HT显著调节的磷酸化位点对应的蛋白质和差异蛋白质的数量。 (k) CCK8分析。(l) 流式细胞分析检测CDK1抑制剂处理后A2780细胞γH2AX、EDU和PI阳性细胞数。(m-o)免疫印迹分析。
02
HT促进dNTPs耗竭,dNTPs耗竭负责复制应激和DNA损伤积累
在CDK1诱导的复制应激和dNTPs饥饿最终导致复制崩溃和细胞死亡后,dNTP稳态在细胞命运中起着重要作用。RRM2是一种核糖核苷酸还原酶亚基,在应对复制应激时,对维持dNTP稳态防止复制灾难至关重要。CDK1磷酸化RRM2 Thr33位点,并通过E3泛素连接酶SCF (Cyclin F)促进RRM2泛素化和降解。因此,他们检测了RRM2的动态表达,发现在OVCAR8、A2780和ID8细胞中,RRM2在高温后逐渐降低(图2a),这与高温后T14和Y15上CDK1磷酸化的降低是同步的(图1e)。
为了证实HT后广泛的复制应激和DNA损伤积累是对CDK1超激活导致核苷酸池耗竭的靶反应,他们推断dNTPs (dNs)的补充可以挽救HT诱导的复制应激和DNA损伤,实验证明,补充dNTP确实减轻了HT诱导的多种癌细胞系的细胞死亡(图2b)。在功能上,外源性dNTPs足以减少HT诱导的非复制S和G2/M细胞(图2c)。此外,补充dNTPs可减轻HT诱导的γH2AX阳性细胞(主要处于非复制S期和G2/M期)的损伤(图2c)。ssDNA和泛核γH2AX染色进一步证实了dNTPs在OVCAR8细胞中显著减弱了HT诱导的复制应激和DNA损伤(图2d)。这些结果共同表明,HT后dNTPs耗竭导致了DNA复制应激和DNA损伤积累。
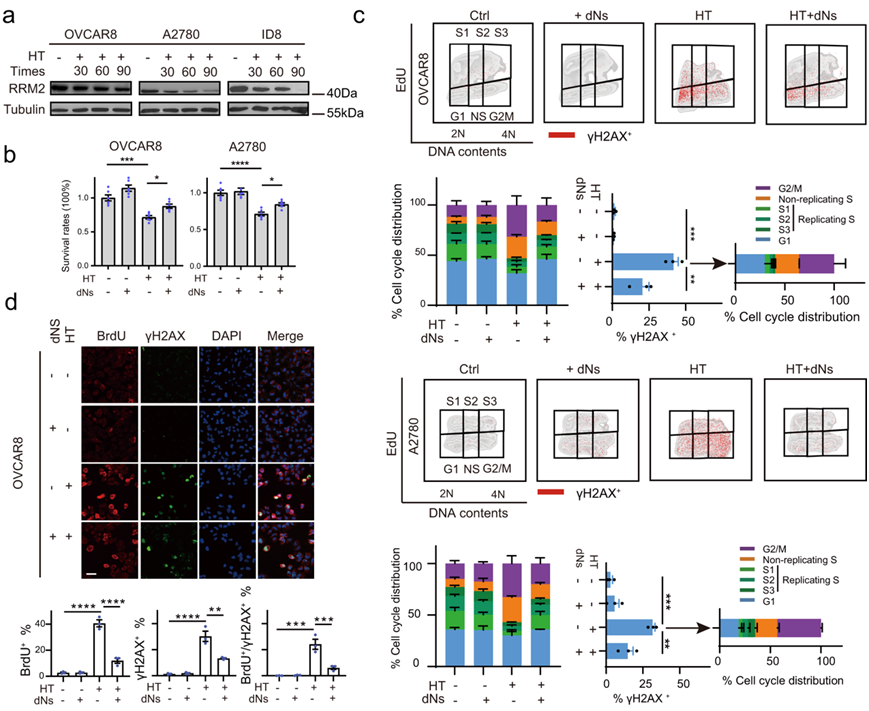
图2. 高温会促进 dNTP 消耗,从而导致复制应激和 DNA 损伤累积。
(a) 免疫印迹分析。(b) 在用 HT 处理 90 分钟之前,将细胞在有或没有 dNTP 混合物 (dNs, 50 μM) 的情况下培养 48 小时,检测细胞活力的变化。(c) 流式细胞分析。(d) BrdU 和 γH2AX 阳性 OVCAR8 细胞的代表性图像和定量。
03
HT后CDK1激活及恢复的动态变化
HT诱导的多种分子事件或生物学通路呈现动态变化。42℃高温处理90 min后,返回37℃后,CDK1活性发生强烈激活,逐渐增加,并持续数小时(OVCAR8、A2780和ID8细胞分别为3、6和24小时以上)(图3a)。值得注意的是,HT诱导的CDK1过度激活似乎是可逆的,尽管恢复时间在不同的细胞中有所不同(OVCAR8、A2780和ID8细胞分别为6、12和超过48小时)(图3a)。令人印象深刻的是,以γH2AX为代表的DNA损伤和以cleaved caspase-3为代表的细胞凋亡随着CDK1活性的变化同步增加和恢复,强烈表明两者之间存在因果关系(图3a)。在ID8细胞中,HT诱导的CDK1激活、DNA损伤和凋亡持续长达48小时,可能是因为这些细胞对HT表现出更高的敏感性(图3a)。
功能上,EdU和γH2AX共标记流式细胞分析显示了高温后的动态复制应激和随后的DNA损伤动力学特征(图3b)。正如预期的那样,OVCAR8和A2780细胞中非复制的S期和G2/M期细胞分别在3小时(h)和6h内持续增加(图3b)。同样,在OVCAR8细胞中,γH2AX阳性细胞逐渐增加,在HT后3 h时达到峰值,在6 h时恢复,主要为非复制S期(表明细胞正在经历复制突变)和G2/M期(细胞仍在经历DNA损伤时过早进入有丝分裂的标志)(图3b-c)。免疫荧光共染色ssDNA和γH2AX证实在OVCAR8、A2780和ID8细胞中存在ssDNA阳性和ssDNA-γH2AX双阳性细胞,分别增加并维持3、6和24小时以上(图3d)。
总的来说,这些结果揭示了高温诱导的CDK1激活、复制应激、DNA损伤以及细胞恢复到正常温度后最终死亡的动态和可逆变化。
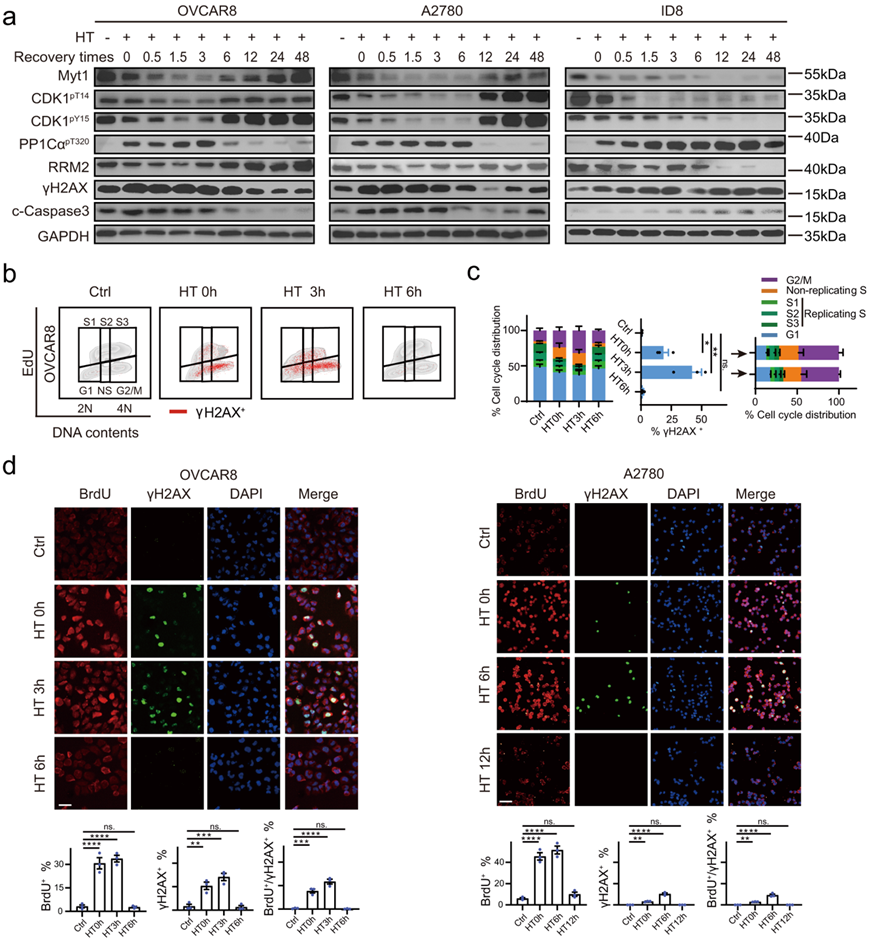
图3. 高温处理后CDK1激活和恢复的动态变化。
(a) 免疫印迹分析。(b-c)流式细胞分析。 (d) BrdU 和 γH2AX 阳性 OVCAR8 和 A2780 细胞的代表性图像和定量。
04
HT通过增加复制应激、DNA损伤和诱导有丝分裂灾难使WEE1抑制剂致敏
癌症治疗中的“组合拳”方法需要连续应用两种不同的治疗或干预措施来提高治疗结局。HT诱导CDK1超激活、G2/M期扰动、复制应激和DNA损伤(图1)。因此,他们在OVCAR8和A2780细胞中分别靶向或利用CDK1相关的G2/M细胞周期检查点、复制应激或DNA损伤通路,通过将HT与15种特性良好的抑制剂联合使用,进行了协同潜力的候选药物筛选(图4a-b)。同时,以HIPEC标准组分(DDP)与紫杉醇(TAX)联合作为阳性对照。
在本文中,他们应用了一种新兴方法来评估HT对单个抑制剂的致敏效应:ΔAUC(拟合剂量-反应曲线下面积)% = [AUC (Ctrl)−AUC (HT)]/AUC (Ctrl)(图4c),较高的ΔAUC%意味着HT和药物之间更大的协同潜力。HT与WEE1i (AZD1775)联合在OVCAR8和A2780细胞中均表现出最一致和最强的协同作用,超过了DDP和TAX的联合作用(图4d)。
WEE1通过磷酸化Y1552抑制CDK1的活性。它还通过依赖CDK1的复制起始点异常激活和非预定的有丝分裂进入诱导复制应激和细胞死亡。因此,他们假设HT和WEE1i之间的协同作用可能依赖于它们对CDK1活性的影响。事实上,与细胞活力和凋亡实验一致,HT和WEE1i的结合增强了CDK1的激活,CDK1-pY15进一步降低,PP1Cα-pT320增加(图4e)。此外,与WEE1i单独治疗相比,HT和WEE1i联合治疗增加了DNA损伤(γH2AX)和细胞凋亡(裂解的PARP)(图4e)。值得注意的是,与图3所示的结果一样,HT单药治疗在OVCAR8和A2780细胞中恢复了对CDK1激活、DNA损伤和凋亡的诱导(图4e)。处理48 h后,他们用EdU、PI和γH2AX对细胞进行标记。正如预期的那样,AZD1775单药治疗适度增加了非复制的S期和G2/M期细胞,而联合治疗进一步增强了这些表型,表明DNA复制应激和G2/M检查点失败(图4f)。γH2AX阳性细胞,主要处于非复制S期和G2/M期,在联合作用下进一步增强(图4f)。此外,虽然AZD1775在处理24小时后显著降低了DNA复制叉的速度,但HT和AZD1775联合使用几乎停止了复制叉的进展(图4g)。在ssDNA和γH2AX共染色的OVCAR8细胞中观察到类似的结果,进一步表明复制应激和随后的DNA损伤(图4h)。此外,虽然WEE1i适度增加了组蛋白H3 Ser10磷酸化(pH3)有丝分裂细胞的比例,但HT和WEE1联合处理抑制大量增加了pH3+细胞的比例(图4i)。最重要的是,HT和WEE1i同时处理显著增加了pH3和γH2AX双阳性细胞的比例,而WEE1i单独处理仅轻微增加了这些双阳性细胞的比例。
与WEE1作为S期和G2/M期检查点的关键介质的作用一致,AZD1775单独处理导致适度的DNA损伤和M细胞积累(图4j-k)。在所有测试的原发性卵巢癌细胞中,AZD1775联合HT进一步加剧了这些事件(图4j-k)。
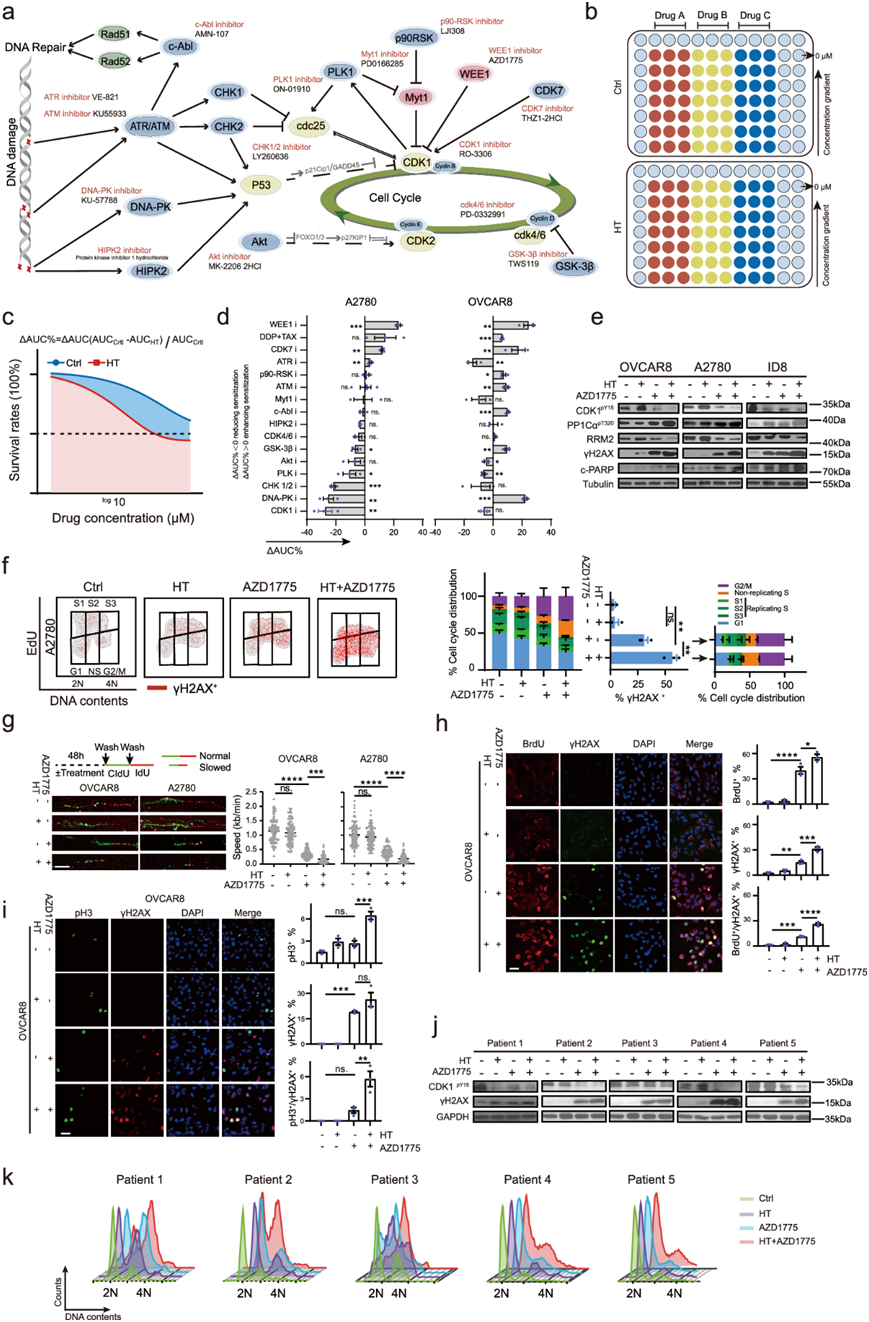
图4. HT通过增加复制应激、恶化 DNA 损伤并诱导有丝分裂灾难来使 WEE1 抑制剂敏感。
(a) 细胞周期相关抑制剂在热疗(HT)协同药物筛选中的功能和商品名称。(b) 药物筛选的流程图。(c) HT对药物的致敏作用定义为ΔAUC% = [AUC(Ctrl)−AUC(HT)]/AUC(Ctrl)。(d) 计算HT与各种药物联合用药的ΔAUC%。(e) 免疫印迹分析。(f) 流式细胞分析。(g) DNA纤维分析。(h) BrdU和γH2AX阳性OVCAR8细胞的代表性图像和定量。(i) pH3和γH2AX阳性OVCAR8细胞的代表性图像和定量。(j)免疫印迹分析。(k)细胞周期流式细胞分析。
05
CDK1过度激活和dNTPs饥饿介导了HT和WEE1抑制剂之间的协同作用
如果CDK1过度激活和dNTPs耗竭(由于CDK1过度激活)是HT和WEE1抑制之间协同作用的原因,那么CDK1抑制应该可以挽救AZD1775和HT之间的合成致死性。事实上,用RO-3306抑制CDK1不仅减轻了AZD1775诱导的细胞死亡,而且还消除了AZD1775与HT之间的合成致死(图5a)。同样,外源性dNTPs的补充也能在较小程度上挽救AZD1775和HT处理的合成致死率(图5a)。在功能上,CDK1的抑制减轻了WEE1i单药治疗和联合治疗诱导的DNA损伤和复制突变的影响(图5b-d),强调了CDK1激活在HT和WEE1i之间的协同作用中发挥的关键作用。
在cdk1诱导的复制应激反应中,dNTP稳态对于防止复制灾难至关重要。因此,通过HT和WEE1抑制CDK1激活的增加可能导致dNTPs水平极低,从而导致复制崩溃和细胞死亡。事实上,dNTPs的补充也消除了HT和WEE1i之间的合成致死相互作用(图5e-g)。这些数据表明,CDK1过度激活和随之而来的dNTPs耗竭至少部分促成了ht诱导的细胞对WEE1抑制的易感性。
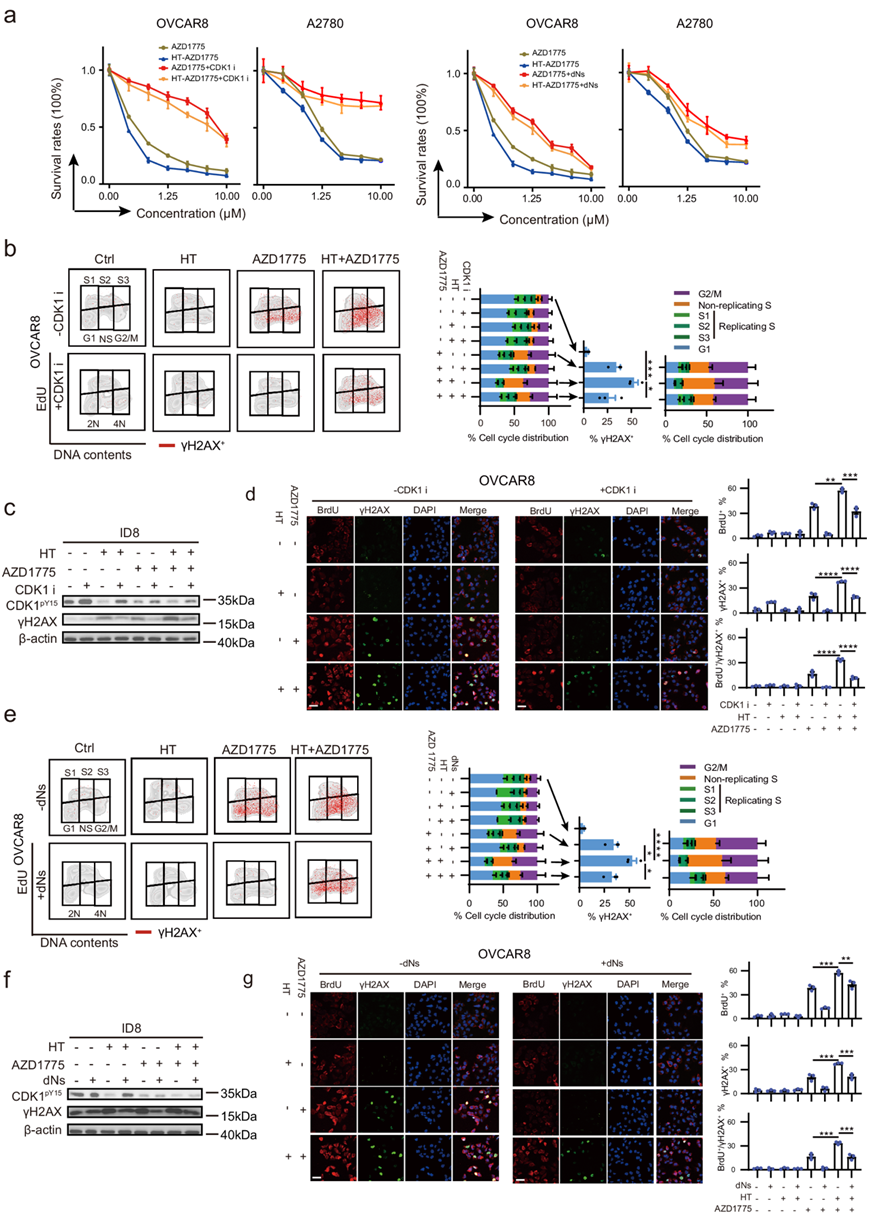
图5. CDK1 过度激活和 dNTP 饥饿介导 HT 和 WEE1 抑制剂之间的协同作用。
(a) 剂量反应曲线分析。(b) 流式细胞分析。(c)免疫印迹分析。(d) BrdU 和 γH2AX 阳性 OVCAR8 细胞的代表性图像和定量。(e)流式细胞分析。(f)免疫印迹分析。(g) BrdU 和 γH2AX 阳性 OVCAR8 细胞的代表性图像和定量。
06
HT和WEE1抑制联合治疗的联合模式策略和治疗窗口
由于他们已经确定 HT 通过复制停滞和有丝分裂灾难选择性增强对 WEE1 抑制的抗肿瘤反应,因此他们将探究这些疗法的施用顺序和组合方式将会是最佳组合策略(图 6a)。有趣的是,无论是同时还是顺序施用,在WEE1i之前施用HT,在杀死OVCAR8、A2780和ID8细胞方面观察到显著的协同效应(图 6b-c)。相反,当给药顺序颠倒时(即 WEE1i 先于 HT),协同作用并不明显。接下来他们探索了 HT 和 WEE1 抑制联合治疗的治疗窗口(图 6d)。与 HT 诱导的各种细胞中 CDK1 动态激活和恢复一致,在 WEE1 抑制之前应用 HT 时观察到序贯组合的协同致死效应,OVCAR8 细胞的最佳治疗窗为 6 小时内,A2780 细胞的最佳治疗窗为 12 小时内。对于ID8细胞,延长至48小时甚至更长(图 6e-f)。这些发现强调了考虑治疗时间和顺序的重要性,以最大限度地发挥 HT 与 WEE1i 组合的协同潜力。重要的是,这些结果强调了以 HT 治疗作为初始干预,然后抑制 WEE1 的重要性,以最大限度地发挥这些细胞系中的协同杀伤作用。
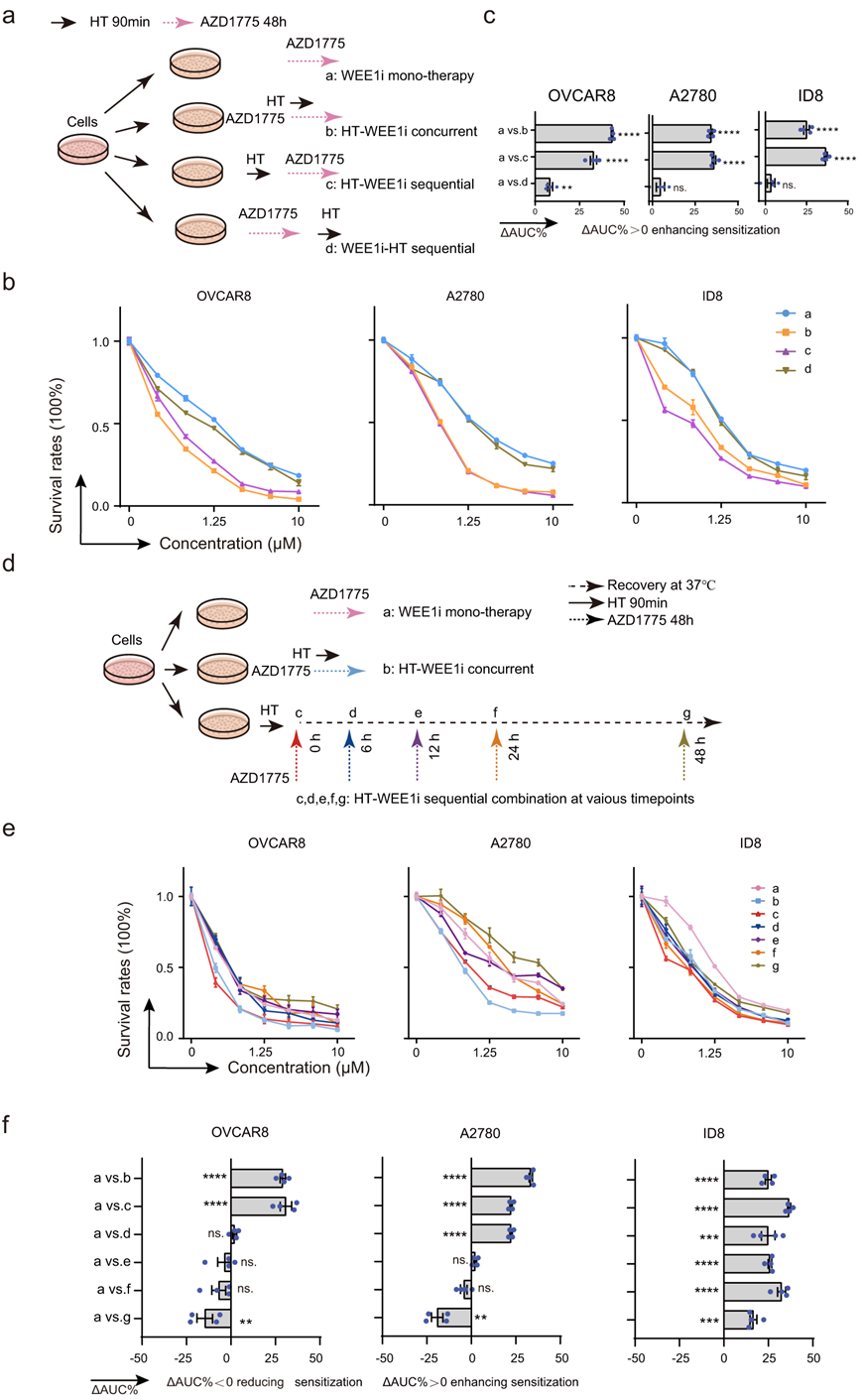
图6. 热疗和WEE1抑制联合治疗的联合模式策略和治疗窗口。
(a) 热疗(HT, 42°C, 90分钟)和WEE1抑制剂(WEE1i, AZD1775)的顺序或并发联合模式策略流程图。 (b-c) 剂量-反应曲线和ΔAUC%分析。(d) HT和WEE1i的并发或多种顺序组合策略流程图。(e-f) 给出了不同组合方式的剂量-反应曲线和ΔAUC%。 (g) (h) (i) (j) (k) (l) (m) (n)
07
Myt1 下调导致热疗诱导的 CDK1 过度激活以及 WEE1 抑制的合成致死率
为了探索HT后CDK1激活的上游分子,他们重新分析了本项研究的蛋白质组学数据,并注意到除了WEE1外,Myt1是另一种抑制性CDK1激酶,在HT后显著下调。WEE1通过磷酸化Y15上的CDK1抑制CDK1的激活,而与WEE1结构相关的Myt1通过抑制T14/Y15上的磷酸化和细胞质中CDK1的隔离来负性调节CDK1。因此,他们推断Myt1下调可能有助于ht诱导的CDK1过度激活,并有助于观察到WEE1抑制的合成致死性。首先,与CDK1-pY15的逐渐减少一致,Myt1和CDK1-pT14(Myt1的另一个下游靶点)在HT后也以时间依赖性的方式下调(图1e)。其次,与CDK1-pY15的动态趋势平行,Myt1和CDK1-pT14在HT后同步降低并恢复(图3a)。更重要的是,Myt1和CDK1-pY15在5个原发性HGSOC培养中观察到动态变化,尽管不同患者的持续时间和恢复时间不同(图7a)。
为了证实Myt1下调是ht介导的CDK1过度激活的主要原因,他们在OVCAR8和ID8细胞中稳定地过表达Myt1,显著增加了抑制CDK1- py15和CDK1- pt14磷酸化(图7b)。此外,Myt1的过表达减少了ht诱导的DNA损伤(通过减少γh2ax积累表明)和ID8和OVCAR8细胞的细胞死亡(图7c-d)。在功能上,Myt1异位表达减少了高温诱导的非复制S期和G2/M期细胞数量(图7e),并减少了高温后的ssDNA和复制突变(图7f)。因此,Myt1下调至少在一定程度上有助于ht诱导的CDK1过度激活、复制应激和DNA损伤。值得注意的是,HT诱导的Myt1下调也独立于HSF1发生(图s1o)。
先前的研究表明,高水平的Myt1与内在的AZD1775抗性有关,代偿性Myt1激活促进了癌细胞对WEE1抑制的获得性抵抗。他们证实Myt1、CDK1-pY15和CDK1-pT14在wee1耐药细胞(ID8R)中显著升高,与亲本细胞(ID8细胞)相比,IC50增加了10倍(图7g-h)。同时,通过siRNA下调Myt1使ID8R细胞对AZD1775敏感(图7i-j)。此外,异位Myt1表达使亲本ID8和OVCAR8细胞对AZD1775产生抗性,这加强了他们的结论,即Myt1是AZD1775敏感性的关键决定因素(图7k)。值得注意的是,HT至少部分降低了Myt1、CDK1-pY15和CDK1-pT14,增强了HT诱导的DNA损伤和凋亡(图7l),甚至在ID8R细胞中也恢复了对AZD1775的敏感性(图7m),证明HT有可能通过下调Myt1来克服获得性WEE1i抗性。
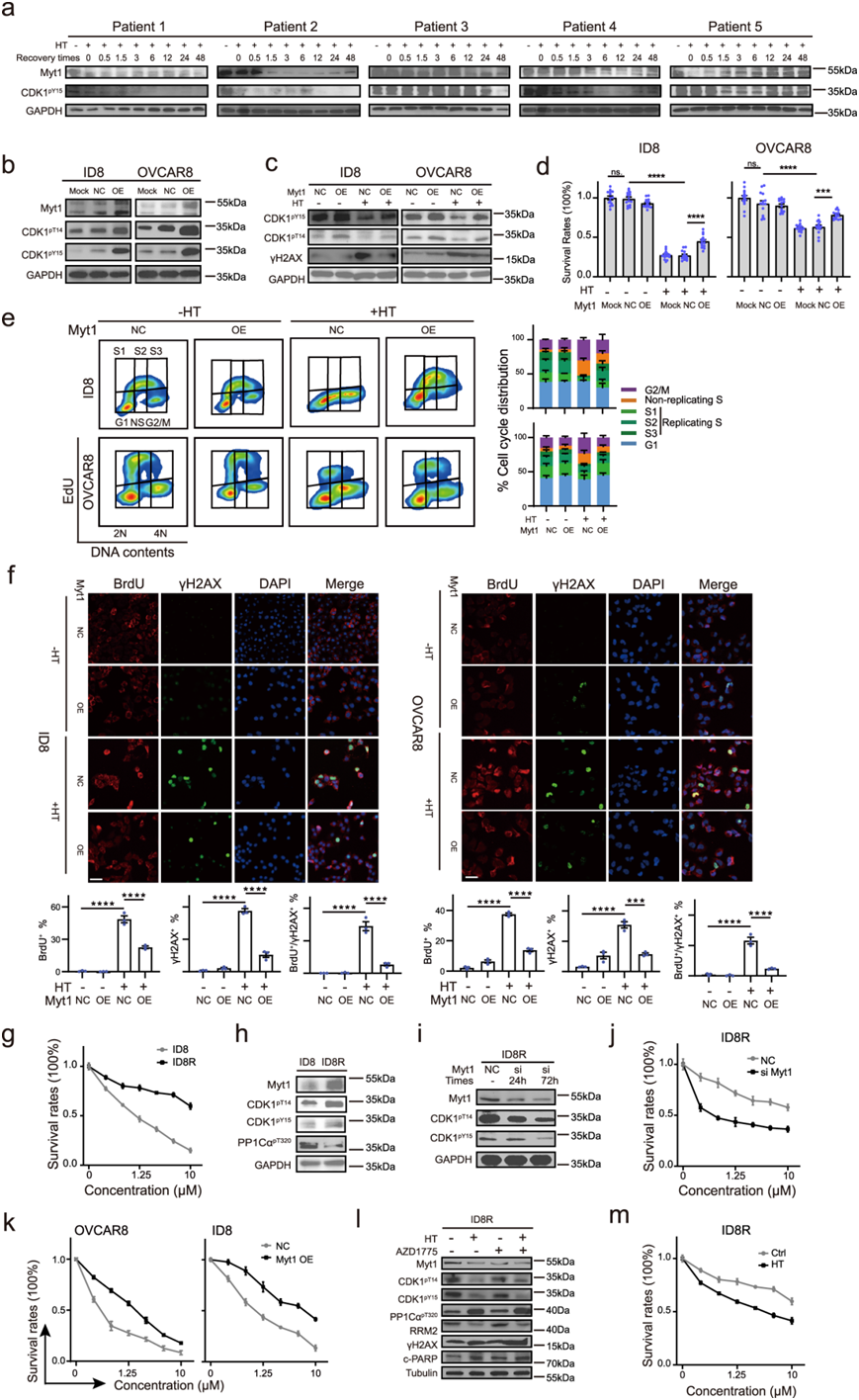
图7. Myt1下调导致热疗诱导的 CDK1 过度激活以及 WEE1 抑制的合成致死率。
(a-c) 免疫印迹分析。(d) 剂量反应曲线。(e)流式细胞分析。(f)免疫荧光分析。(g) 用递增剂量的 AZD1775处理 48 小时的 ID8 和 ID8R 细胞中细胞活力的剂量反应曲线。(h-i) 免疫印迹分析。(j-k) 剂量反应曲线。(l)免疫印迹分析。(m)剂量反应曲线。
08
腹腔热灌注和WEE1i在体内对卵巢肿瘤有明显的消退作用
他们的体外实验提供了关于HT和WEE1抑制联合治疗对卵巢癌细胞的协同作用的有趣发现。因此,他们开发了一种模仿临床治疗方案的小型化、简化且可重复的装置,以验证 HIPET 与 WEE1i 对小鼠的体内疗效。该闭路设备由控温水浴、灌注液储备瓶、流入/流出导管、滚子泵和温度检测和显示系统组成(图8a)。
通过将荧光素酶转染的鼠ID8卵巢癌细胞系(ID8-luc)腹腔注射到C57BL/6小鼠体内,建立粟粒性腹膜扩散的卵巢癌小鼠模型。一旦用体内成像系统 (IVIS) 检测到肿瘤,将20只小鼠随机分为四组(图 8b)。值得注意的是,在他们评估荷瘤 C57BL/6 小鼠对 HIPET 耐受性的初步试验中,他们发现 30 分钟的 HIPET 耐受性良好,并发症最少。然而,持续时间超过 30 分钟会导致严重的健康问题,包括潜在的死亡。幸运的是,由于ID8对HT的高度敏感性(图 3a),他们检测了不同持续时间的HT刺激(30、60和90分钟)对ID8细胞的影响。因此,他们分别在37°C和42°C条件下以生理盐水溶液连续灌胃小鼠作为对照组和以AZD1775 (60 mg/kg)为治疗组(四组)。术后恢复期24 h后,两组对照组给予载药(2% DMSO + 30% PEG300 + 5% Tween 80 + ddH2O,灌胃)。同时,两治疗组再给予AZD1775 (60 mg/kg/d,口服灌胃,开5天,停2天)21天,或直到对照组出现严重腹水需要终止(图8b)。经过设备优化和程序调整,HIPET过程中及围手术期均无小鼠死亡,全部存活至研究终点;随后,进行IVIS,收获肿瘤组织,并通过免疫组化(IHC)评估其形态学、增殖、DNA损伤和CDK1激活。与ID8细胞在体外对HT的高敏感性一致,与正常灌注的对照动物相比,IVIS显示,30分钟HT输注适度降低肿瘤负荷(图8c-d),腹围显示腹水(图8e)。同时,AZD1775单药治疗比单纯HT治疗更能减轻肿瘤负担(图8c-e)。将WEE1i与HT联合腹腔注射后,肿瘤明显消退(图8c-e)。
正如预期的那样,研究结束时对肿瘤的免疫结构分析表明,在WEE1i治疗的肿瘤中,CDK-pY15大量减少,这与WEE1i在整个研究期间使用的剂量下完全抑制其靶标一致(图8f)。值得注意的是,HT和WEE1i联合使用进一步强化了这种减少,表明协同治疗组发生了CDK1超激活(图8f)。在HT和WEE1i单独治疗的肿瘤中,指示增殖的Ki67轻微下降,而在HT/WEE1i联合治疗的小鼠中,Ki67大量下降。这伴随着γh2ax的诱导,表明在HT或WEE1i治疗的肿瘤中DNA损伤,特别是在接受HT/WEE1i联合治疗的小鼠中(图8f)。
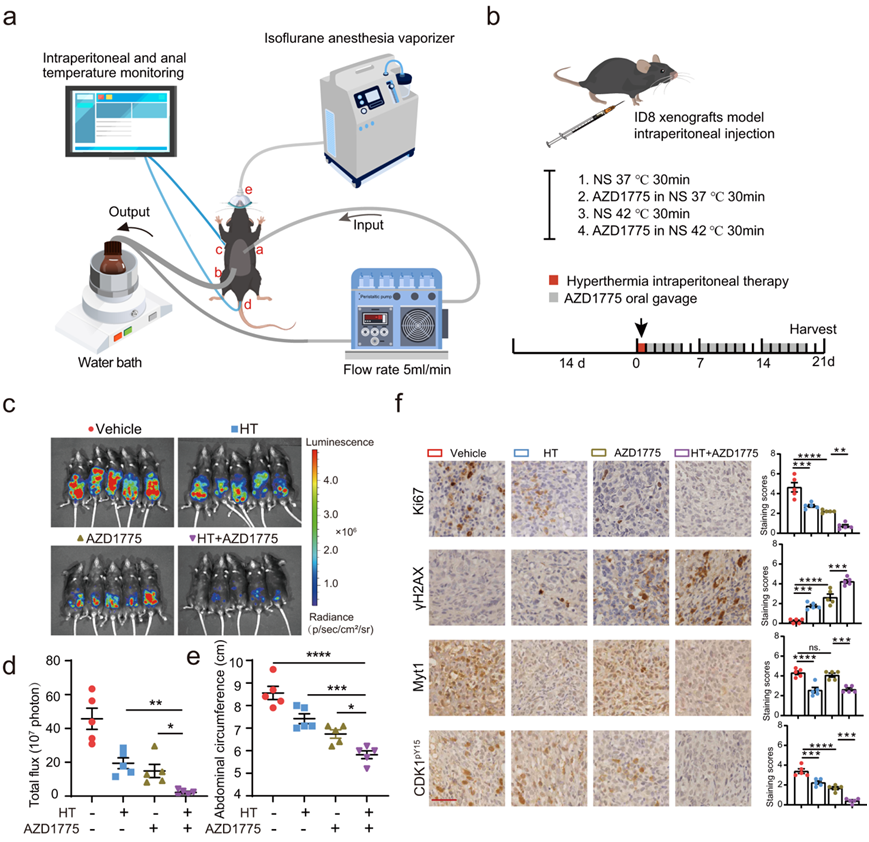
图8. 腹腔热灌注和 WEE1i 可显著消退体内卵巢肿瘤。
(a-b) 小鼠腹腔热疗 (HIPET) 设置的示意图。(c) 持续监测腹膜内和肛门温度。 (d-e) C57BL/6小鼠生物发光的定量。 (f)组织学分析。
+ + + + + + + + + + +
结 论
本项研究对热疗下的卵巢癌细胞进行了全面的多组学研究,揭示了独特的分子全景,主要特征是快速的蛋白质磷酸化变化。根据磷酸化特征,确定 CDK1 激酶在高温期间过度激活,从而影响整体信号传导格局。他们观察到动态、可逆的 CDK1 活性,导致复制停滞和高温后早期有丝分裂进入。随后的药物筛选表明,WEE1 抑制可与热疗协同破坏癌细胞。内部开发的小型装置证实热疗和 WEE1 抑制剂组合可显著减少体内肿瘤。这些发现为 HIPET 提供了更多见解,详细说明了热疗的分子机制并确定了用于靶向治疗的精确药物组合。这项研究推动了精确腹腔热腹治疗的概念,凸显了其对抗卵巢癌的潜力。
+ + + + +



