

 English
English文献解读|Cancer Cell(50.3):空间分辨多组学破译胶质母细胞瘤中双向肿瘤-宿主相互依赖性
✦ +
+
论文ID
原名:Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma
译名:空间分辨多组学破译胶质母细胞瘤中双向肿瘤-宿主相互依赖性
期刊:Cancer Cell
影响因子:50.3
发表时间:2022.06.13
DOI号:10.1016/j.ccell.2022.05.009
背 景
胶质母细胞瘤是中枢神经系统的恶性肿瘤,其特点是亚克隆多样性和发育层次中的动态适应,然而这些肿瘤空间背景下动态重组的分子机制仍然是未知的。
实验设计
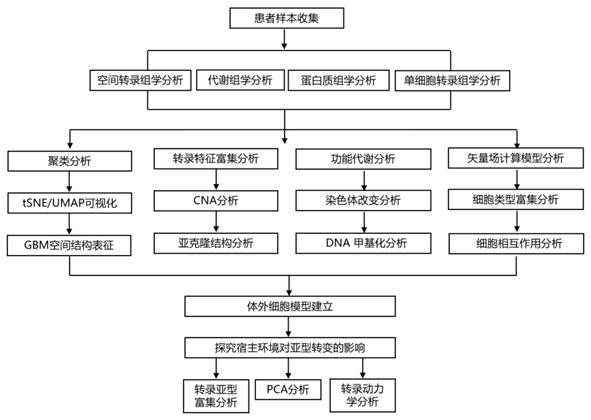
结 果
01

胶质母细胞瘤空间分辨转录组学图谱
为了表征胶质母细胞瘤(GBM)的空间结构,研究者团队整理了28个样本的空间转录组(stRNA-seq)图谱,得到了88793个不同年龄组和解剖区域的转录组数据。他们补充了空间代谢组学和蛋白质组学分析,然后用于stRNA-seq,以全面整合多个分子层(图1A)。基于互近邻(MNN)的水平整合和共有近邻(SNN)聚类显示,非恶性标本(通路皮层)在患者之间表现出相似性。恶性转录组具有独特的基因表达谱,恶性来源的样本熵值明显降低,表明该聚类是由单个患者的斑点组成的(图1B-C)。在所有样本的 88793 个斑点中,有63121 个斑点来自恶性样本,其中 46459 个斑点至少含有 95% 的肿瘤细胞(图 1 C)。为了将本项的结果与组织学分类的现行金标准相结合,他们根据 Ivy GAP 组织学分类系统预测了点状组织学表型(图 1D)。与组织病理学特征相比,肿瘤细胞频率低的样本主要包含浸润区域(图1E)。
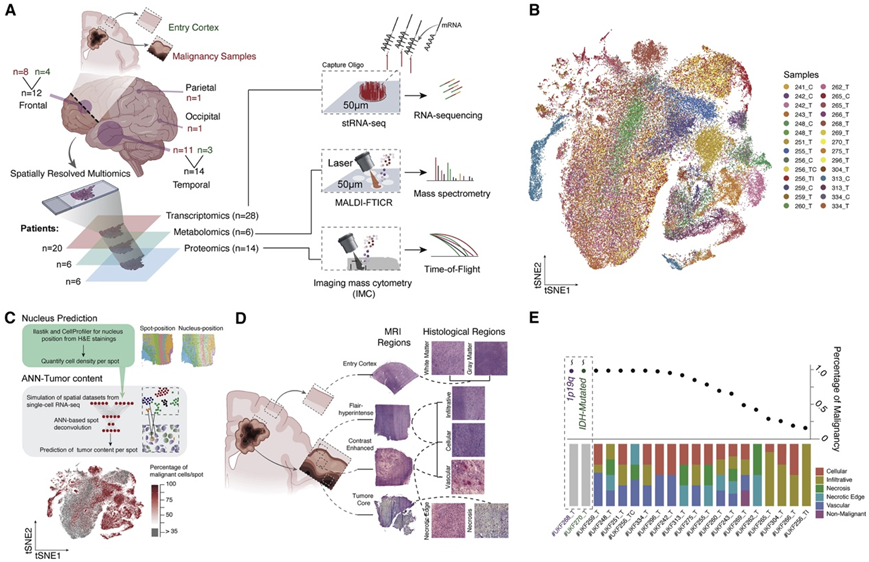
图1. 方法和队列概述。
(A) 工作流程和空间数据集群组的图示(左)以及所使用的分析方法的概述(右)。(B) 所有集成的空间分辨转录组的斑点 t 随机邻域嵌入 (tSNE) 图。(C) 肿瘤细胞含量预测的实验流程概述(上)和 tSNE 图(下)。(D) 组织学定义区域的不同分辨率的示例。(E) 基于人工神经网络(ANN)预测的 stRNA-seq 数据集中恶性斑点百分比的点图。
02
破译空间分辨的转录异质性
在转录空间内,他们发现了5个一致表达的转录程序,后来称为“空间上不同的转录程序”(图2A)。在笛卡尔空间中,他们随后整合了患者的空间加权相关矩阵,然后进行了层次聚类,确认了五个反复出现的空间上不同的转录程序的空间分离(图2A)。他们发现了两个空间上不同的转录程序,它们与神经胶质相关基因(例如GFAP、AQP4、VIM、CD44)的高表达相关。虽然这两个程序都代表神经胶质谱系,但一个程序与放射状神经胶质相关基因(HOPX、PTPRZ1)的表达增加有关,另一个显示炎症相关基因(如HLA-DRA、C3、CCL4、CCL3)和基因集的功能富集。这些具有共同神经胶质转录谱的程序将分别进一步称为“放射状神经胶质”和“反应性免疫”(图2A)。其余的转录程序显示了与神经胶质谱系的基本一致性,并根据其神经(称为“神经发育”)或少突胶质细胞起源(称为“空间OPC”)(图2B-C)。第五个空间上不同的转录程序,进一步称为“反应性缺氧”,与缺氧反应(如VEGFR、HMOX1、GAPDH)和糖酵解(如LDHA、PGK1)基因有关,这表明代谢改变加上低浓度的氧气驱动一些区域的不同转录状态(图2B-C)。
他们假设这种表型的特点是动态适应反应,因此与微环境的不同变化有关。
反应性缺氧程序表现出与类间充质(MES)样亚型最强的重叠,特别是缺氧依赖性“MES2”状态(图2C)。在反应性缺氧相关斑点中,他们发现拷贝数改变(CAN)显著积累,作为独立的亚克隆事件发生(图2D)。
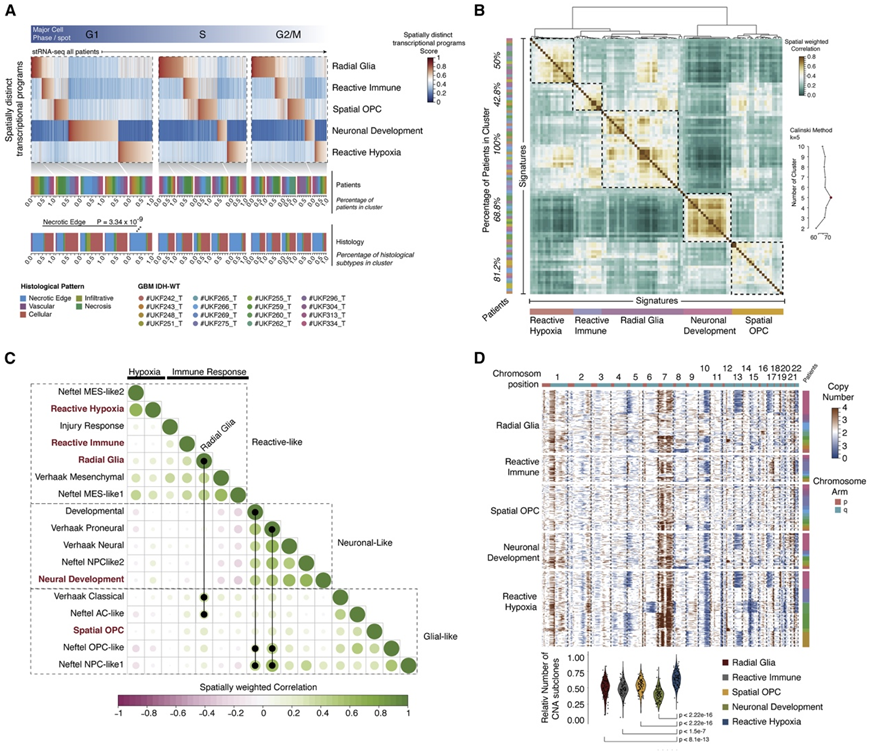
图2. 空间上不同的转录程序的探索。
(A) 胶质母细胞瘤空间分辨转录组学的热图。(B) 99 个单独转录程序的空间加权相关分析的热图。(C) 当前转录特征的富集分数和空间重叠的空间加权相关分析。(D) 不同 CNA 跨空间不同转录亚型的热图。
03
空间上不同的转录程序和亚克隆结构
接下来,他们的目的是研究空间上不同的转录多样性是否可以直接反映肿瘤内的遗传亚克隆。他们通过患者特异性的 CNA 层次聚类重建了克隆结构。鉴定出 57 个遗传亚克隆,每个样本有 2 至 6 个亚克隆(图 3 A)。然后他们确定了每个亚克隆内个体、空间上不同的转录程序的分布(图 3B-C)。在 26.32% 的所有亚克隆中,单一转录程序占主导地位(每个亚克隆超过 75% 的斑点)。克隆结构对空间上不同的转录程序的发生影响有限,尽管亚克隆偶尔偏向于空间 OPC或反应性缺氧程序(图 3D )。
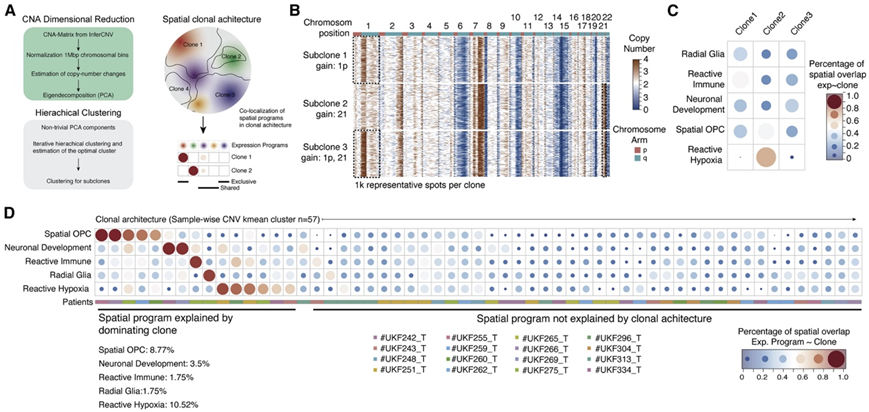
图3. 转录亚群独立于亚克隆架构。
(A) 实验流程概述(左)以及亚克隆内空间结构和转录程序概念的说明。(B) 样本 #UKF 260_T 的 CNA 热图显示 3 个主要亚克隆。(C) 点图表明来自示例 #UKF 260_T 的转录亚组(行)和亚克隆(列)之间的空间重叠。(D) 点图表明所有亚克隆的转录亚组(行)和亚克隆(列)之间的空间重叠。
04
与 GBM 反应性缺氧程序相关的代谢改变
为了进一步探索空间上不同的反应性缺氧程序,他们使用 MALDI 傅里叶变换离子回旋共振成像质谱 (MALDI-FTICR-MSI) 对与用于 stRNA-seq 的组织切片连续的组织切片进行了空间分辨代谢组学分析(图 4 A)。他们实施了类似于 stRNA-seq 所描述的破译代谢谱的工作流程,包括三个主要步骤:(1) 代谢和转录组数据的比对,(2) 患者探索,以及 (3) 相互主成分分析 (RPCA) 。降维揭示了前两个主成分之间的主要差异,从而在校正聚类稳定性后确定了三个突出的代谢亚群(图4A-B)。功能代谢分析显示,在第一个代谢模块(M-G1)中戊糖磷酸途径显著富集。第二个代谢亚群(M-G2)的特点是磷酸腺苷酸代谢富集,是胶质瘤代谢的标志。最终代谢亚群(M-G3)强烈富集糖酵解和氨基糖代谢(图4C)。
在组织学上,反应性缺氧程序的大多数斑点代表坏死边缘(图 4 C)。他们假设缺氧代谢导致空间隔离的生态位中基因组不稳定性的积累。这些“反应岛”代表了从头基因组改变的潜在来源,有助于肿瘤细胞治疗耐药性的进化。
为了验证他们的假设,他们探索了富含反应性缺氧特征的区域。这些区域定义为缺氧核心(双侧缺氧:MALDI 和 stRNA-seq)或缺氧相关区域(单侧缺氧 MALDI 或 stRNA-seq),并富集糖酵解途径(图4D)。与随机选择的对照斑点相比,跨缺氧核心和缺氧相关区域的 CNA 图显示位于缺氧核心区域的染色体 15p和 14q的缺失,而7号染色体显著增益。一些样本还显示出多个染色体(8p、9p、13q、19q和21q)的单一缺失和增益,从而证实了与缺氧相关的代谢是基因组不稳定的潜在驱动因素的假设(图4E)。
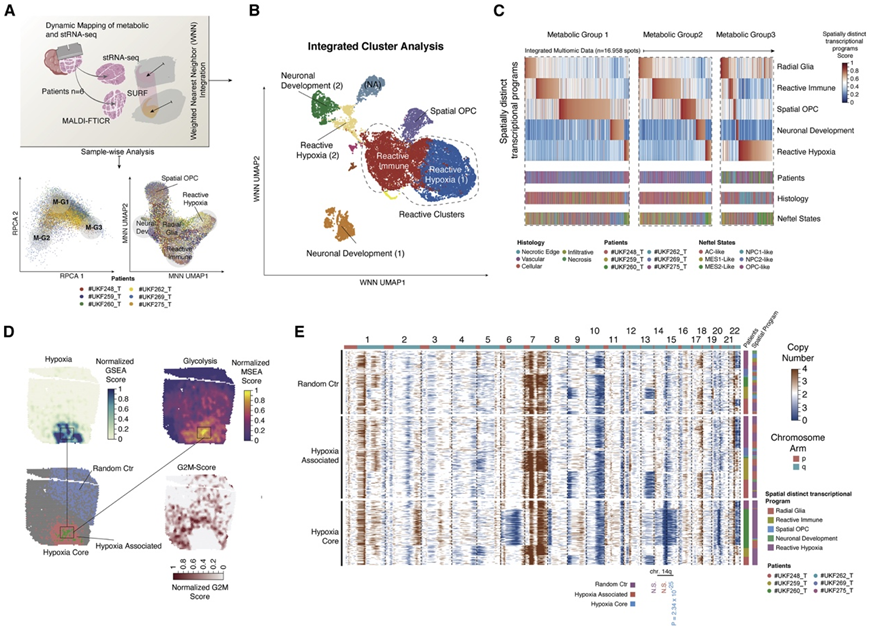
图4. 空间解析代谢组和转录数据的整合。
(A) 整合 6 名患者的转录和代谢组数据的实验流程。(B)使用PCA (RPCA 1-2)对患者代谢数据进行水平整合的散点图,以及使用互近邻(MNN)整合(MNN均匀流形近似和投影[UMAP] 1-2)对转录组数据进行水平整合的散点图。(C)所有整合点的空间分解转录组学热图。(D) 样品 #UKF 275_T 中集成分析的曲面图。(E) 缺氧表型的拷贝数热图。
05
反应性缺氧区域积累染色体改变
为了进一步验证他们的发现,他们分析了来自 TCGA 数据库的样本(GBM IDH1/2野生型),根据缺氧基因表达评分对患者进行分类(图 5 A-B)。缺氧驱动的肿瘤显示染色体改变显著增加,进一步证明了代谢与基因组不稳定性之间的关系(图5C-D)。接下来,他们在常氧和缺氧条件下培养原代患者来源的 GBM 细胞系 2-6 周(图 5E)。
代谢表观遗传和遗传变化之间的具有深刻的联系,DNA 甲基化疾病与 CNA相关。考虑到这一点,他们通过分析常氧和缺氧条件下 CpG 位点 cg12434587 和 cg12981137 的 O-6-甲基鸟嘌呤-DNA 甲基转移酶 (MGMT) 启动子甲基化,探讨了缺氧代谢对 DNA 甲基化的影响。在常氧条件下具有未甲基化 MGMT 启动子的细胞系在暴露于缺氧条件时会发生过度甲基化(图 5 F),这些发现进一步验证了关于代谢驱动的 CNA 的假设。
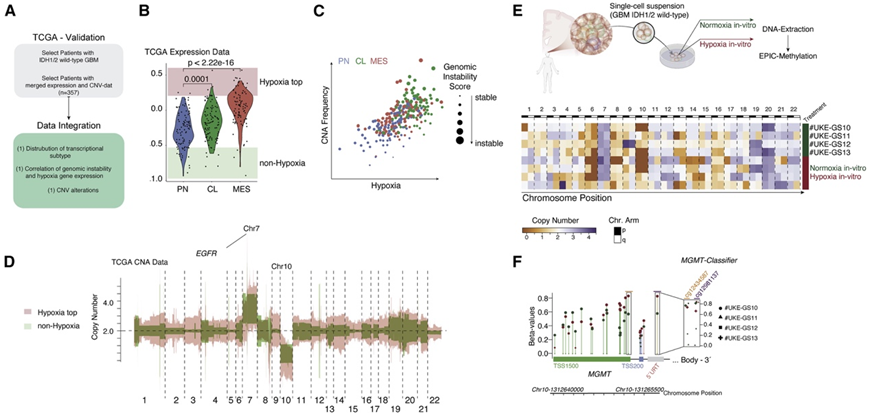
图5. 缺氧相关 CNA 改变的 TCGA 和细胞培养验证。
(A) TCGA 数据分析的概述和实验流程。(B) TCGA 数据验证。(C) CNA 频率和缺氧评分的散点图。(D) 全基因组中 TCGA 数据的 CNA 映射。(E) 在低氧或常氧条件下培养的 4 名患者的原代细胞培养物的实验验证。(F) MGMT TSS1500 处的 CpG 岛–第一个外显子。y 轴显示甲基化β值。lollipop图显示了 MGMT 启动子处缺氧应激反应之间的差异甲基化。
06
缺氧代谢调节 GBM 的“生长或生长”潜力
在对空间分辨转录程序的初步表征中,反应性缺氧程序在非循环细胞中显著富集(图2A)。这与最近的报告一致,表明缺氧应激会引发细胞周期停滞,特别是 S 期停滞(图6A)。这一观察结果在缺氧核心点中很明显,从而引发了新的假设:缺氧驱动的 S 期停滞有助于基因组不稳定性的积累(图 6 A)。
GBM 细胞的基线转录状态可以分为“发育”[类星形胶质细胞(AC)、类少突胶质细胞前体细胞(OPC)或类神经祖细胞(NPC)]样程序。在空间上,这些程序与较低的细胞密度有关,代谢谱显示戊糖磷酸途径(PPP)的富集。由于增殖和肿瘤生长,营养和氧气缺乏不断增加,迫使采用糖酵解代谢程序。这种现象是 PPP 和糖酵解之间的相互转换,与肿瘤细胞的生长或生长潜力有关,表明 PPP 在由生长的肿瘤细胞组成的区域中占主导地位。
为了探索代谢改变区域中迁移基因表达特征的富集,他们确定了特定基因表达特征的低富集和高富集之间的定向梯度的空间方向。简而言之,每个点的方向向量基于其局部邻域中所研究的基因表达特征的梯度富集。这些矢量场计算使得能够近似空间基因表达轨迹,从而能够识别空间相反的转录途径(图 6 B)。基于这些矢量场计算,缺氧反应和迁移特征显示反向空间轨迹(图 6 C-D)。总之,这些结果证明代谢变化和氧化应激是基因组多样性的潜在相互驱动因素,导致 GBM 的克隆进化。
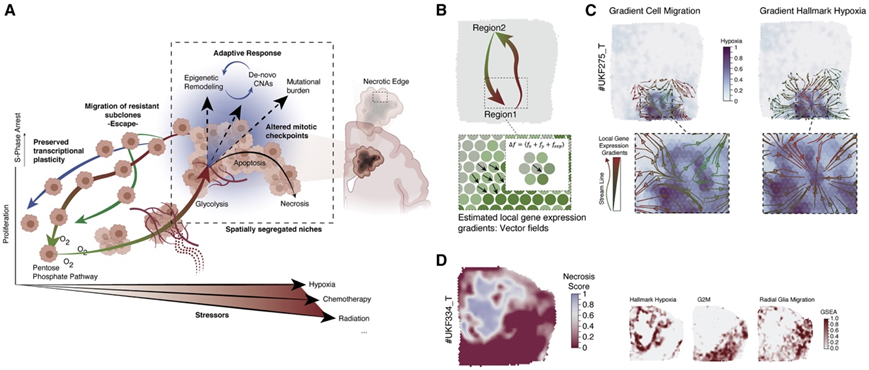
图6. 缺氧应激概念及逃逸机制图示。
(A)增殖-缺氧相互依赖假说的概念和模型。(B) 矢量场计算模型,由基因表达水平空间变化的对齐梯度组成,并指示迁移激活的方向。(C) 空间转录组示例的曲面图 (#UKF275_T)。箭头表示来自定义的转录程序的向量场(显示为流线)。(D) 表面图 (#UKF334_T) 指示坏死(左图)与缺氧富集(左起第二个)、细胞周期(G2M 左起第三个)和放射状胶质细胞迁移(左起最后一个)之间的关系。
07
反应性免疫区域肿瘤与宿主相互依赖性的探索
除了导致空间隔离的缺氧相关环境压力源之外,GBM 的另一个主要压力源是免疫环境,了解免疫环境对于成功开发免疫疗法至关重要。肿瘤内的免疫编辑绝对是一个重要的特征,它确保免疫逃逸并导致 GBM 缺乏有效的免疫治疗选择。反应性免疫分类由富含 MES 样 (MES1) 和星形细胞样 (AC) 转录特征的细胞组成,也称为“MES-AC 混合体”状态(图2B)。
基于成像质谱流式细胞分析(IMC),他们生成82179 个细胞的全蛋白质组图(图 7 A)。在为反应性免疫的区域,髓细胞和淋巴细胞显著增加(图7B-C)。为了研究免疫富集区肿瘤细胞的分化,他们通过将细胞细分为径向胶质细胞(EGFR+HOPX+)、反应性免疫细胞(EGFR+CHI3L1+VIM+)、空间OPC细胞(EGFR+OLIG1+)、神经发育细胞(EGFR+SNAP25+CALM2+)和反应性缺氧细胞(EGFR+VEGFA+)来量化细胞多样性。仅在转录定义的反应性免疫中证实了反应性免疫(EGFR+CHI3L1+VIM+)细胞的独特富集(图7B)。
为了验证关于免疫系统和 GBM 之间的细胞关系,他们建立细胞相互依赖性模型。根据肿瘤细胞与淋巴或骨髓细胞之间的距离量化了它们之间的细胞连接性,证实了肿瘤细胞与转录定义的反应性免疫区域中的免疫区室之间的细胞相互作用增强(图7D)。OPC样细胞主要富集于神经发育区域(图7E-F)。此外,反应性免疫区域和缺氧区域均显示出肿瘤相关骨髓细胞 (TAM) 和 T 细胞的显著富集(图7G)。代表 GBM 免疫景观的 scRNA-seq 数据集的点状投影也证实了肿瘤反应性免疫区域中记忆 T 细胞和耗竭 T 细胞的富集(图 7 H)。
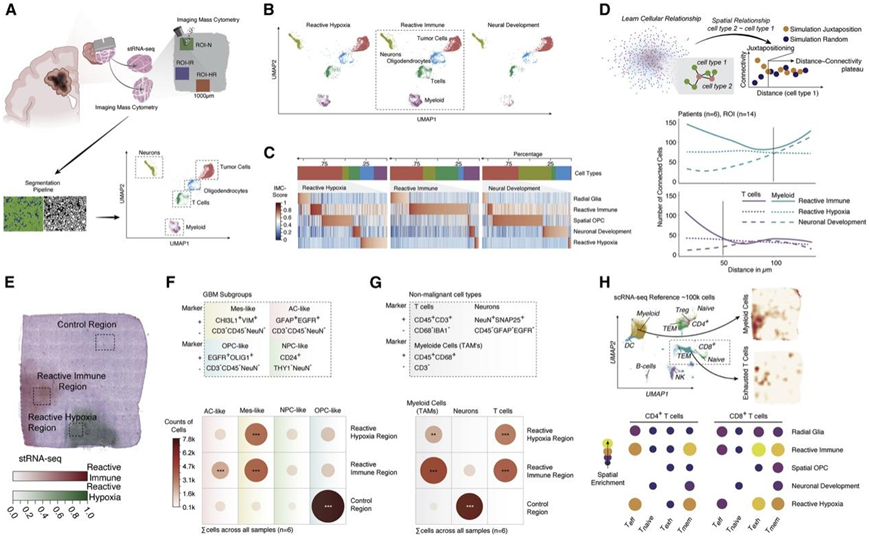
图7. 单细胞质谱流式分析仪的整合。
(A) 成像质量细胞计数数据在空间转录组数据中整合的图示。(B) 主导空间解析转录模式的不同区域中细胞类型的UMAP可视化。(C) 淋巴和骨髓细胞在空间解析转录模式中变化的条形图(上)。注释的空间分辨亚型的肿瘤细胞多样性热图(下)。(D) 细胞关系量化工作流程图示(上)。肿瘤细胞与骨髓(青色)或淋巴(紫色)细胞之间的细胞连接性的量化(下)。(E) 以 #UKF275 的 H&E 为例,表示不同的反应性免疫(红色)和缺氧(绿色)区域。(F-G) 每个分区和细胞类型的细胞计数图示。(H) 以骨髓细胞(右上)和效应 T 细胞的细胞位置反卷积为例,胶质母细胞瘤免疫区室的UMAP可视化(上),CD4+和 CD8+ T 细胞亚型的空间富集热图(下)。
08
环境条件导致双向亚型转变
为了评估各种微环境的影响,他们使用了来自不同年龄(2-85岁)的多个人类供体和来自2周龄和2岁大鼠的两个小鼠供体的皮质通路组织(图8A)。将组织培养4天,之后将相同的mes样原代患者来源细胞系(通过RNA-seq表征)接种到所有培养的组织切片中(图8A-B)。在不同宿主环境中培养7天后,消化组织并进行单细胞转录组分析(scRNA-seq)(15034个细胞)。利用推断的CNA,根据7号染色体的特征增益对肿瘤细胞进行计算鉴定(图8C-D)。
为了探索不同宿主环境下的动态适应性,他们根据其相应细胞状态的富集程度对所有恶性细胞进行了排列(图8E-F)。基线状态(在二维[2D]细胞培养中生长的细胞)显示出mes-1/2样表型,具有低程度的转录多样性和大量循环细胞(图8E)。为了检验动态适应性,他们根据前两个主要成分标注了scRNA速率(图8G)。总的来说,从体外细胞培养的“root(根)”到MES-和ac-样“tail(尾)”的向量场代表了神经环境驱动的定向适应(图8G-i)。蛋白酪氨酸磷酸酶受体类型Z1 (PTPRZ1)是径向胶质细胞分化的GBM细胞的共同标记基因,在Mes - ac 混合状态下恰好处于高速状态(图8G-I)。编码钾通道 (KCNH8) 和代谢型谷氨酸受体 ( GRM3 ) 等基因在这些转录模块中表现出高速状态,表明神经元环境促进确定的转录程序(图8I)。
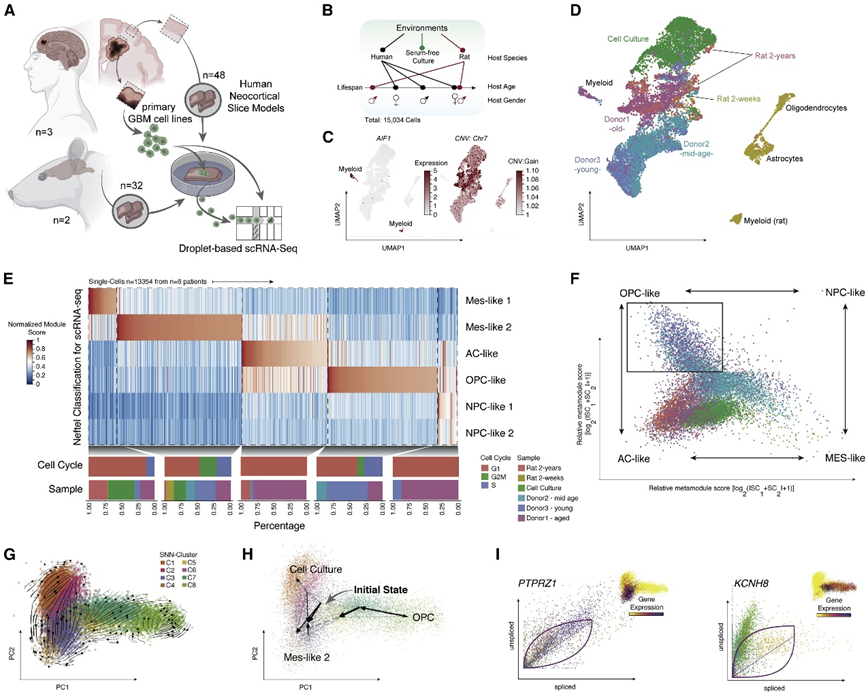
图8. 离体人类皮质培养表明环境影响的双向转变。
(A) 测试不同宿主环境对肿瘤细胞影响的工作流程图示。(B) 宿主环境的概述。(C) 由 AIF1(骨髓细胞)和 7 号染色体(肿瘤细胞)中的增益标记的 scRNA-seq 的降维 UMAP 2D 表示。(D) 所有集成 scRNA-seq 数据的降维 UMAP 表示。(E) 转录亚型的富集分析。(F)四种主要细胞状态的火山图。(G)通过速度流线可视化的主要基因平均流量对应于注射到不同组织供体内后的肿瘤细胞分化。(H) 使用 CellRank 以类似于 (G) 的表示形式估计初始和最终状态。(I) 未剪接基因表达(y 轴)和剪接基因表达(x 轴)的散点图展示了不同聚类内剪接动力学的转录动力学。
+ + + + + + + + + + +
结 论
本项研究通过空间转录组学、代谢组学和蛋白质组学来表征胶质母细胞瘤。通过破译患者之间区域共有的转录程序,推断胶质母细胞瘤是通过谱系状态进行空间分离,并适应炎症和/或代谢刺激。代谢成像和成像质谱流式细胞分析数据的整合揭示了局部肿瘤-宿主的相互依赖性,从而产生了空间排他性的适应性转录程序。推断拷贝数改变强调了与反应性转录程序相关的亚克隆的空间内聚组织,证实环境压力会产生选择压力。模拟各种环境的胶质母细胞瘤干细胞植入人类和啮齿动物新皮质组织的模型证实转录状态源于对各种环境的动态适应。
+ + + + +



