

 English
English文献解读|Microbiome(15.5):肠易激综合征及其排便习惯亚型肠道微生物组的多组学特征
✦ +
+
论文ID
原名:Multi-omics profiles of the intestinal microbiome in irritable bowel syndrome and its bowel habit subtypes
译名:肠易激综合征及其排便习惯亚型肠道微生物组的多组学特征
期刊:Microbiome
影响因子:15.5
发表时间:2023.01.10
DOI号:10.1186/s40168-022-01450-5
背 景
肠易激综合症(IBS)是一种常见的胃肠道疾病,与肠道微生物组的改变有关,但微生物特征一直难以识别。与炎症性肠病等其他胃肠道疾病不同,尚未出现用于 IBS 诊断的可靠的成分或功能微生物组特征。
实验设计
结 果
01

IBS 与微生物分类群、转录本和代谢物的变化有关
本项研究纳入了495 名受试者,包括 318 名 IBS 患者和 177 名健康对照 (HC),并收集了他们的粪便样本。IBS 患者的内脏敏感性 (VSI) 和压力敏感躯体症状 (PILL) 得分明显更高。大约相同数量的 IBS 患者以腹泻为主(IBS-D,38%)和以便秘为主(IBS-C,35%),多数患者症状严重程度为中度(IBS-SSS为175-300,49%)或重度(IBS-SSS为301-500,27%)。
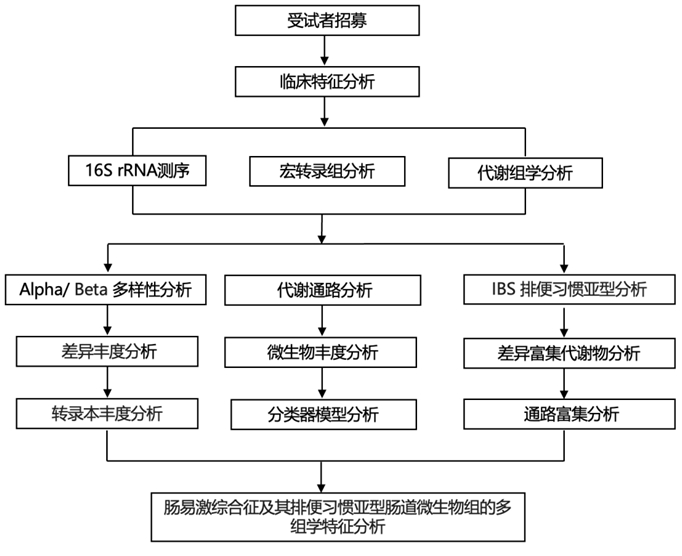
粪便样本通过16S rRNA基因测序、宏转录组测序和非靶向整体代谢组学进行分析。微生物组成通过16S rRNA基因丰度和测序转录本的分类分配来评估。利用Shannon丰富度和均匀度指数比较IBS和HC之间的Alpha多样性(即样本内多样性)。通过非参数检验和多元线性模型,两组数据集的Shannon指数均无显著差异(图1A)。相比之下,使用布雷-柯蒂斯差异进行的Beta多样性分析(即样本间的多样性)显示,IBS和HC之间的16S rRNA谱和宏转录组学分类在组成上存在统计学显著差异(图1B)。鉴于焦虑和抑郁之间存在很强的相关性,并且医院焦虑和抑郁 (HAD)评分在 IBS 和 HC 之间存在较大差异,因此选择此作为协变量来代表情绪与微生物组的关联。在调整这些协变量的多变量分析中,通过 16S rRNA 测序和宏转录组分析,IBS 仍然与微生物组成显著相关,包括年龄和种族在内的几个协变量也是如此(图 1 B-C)。
然后,在调整通过Beta多样性分析确定的影响微生物组的协变量(包括年龄、性别、种族、BMI、饮食和HAD焦虑)后,使用多变量一般线性模型来识别IBS与HC中显著差异的微生物分类群。
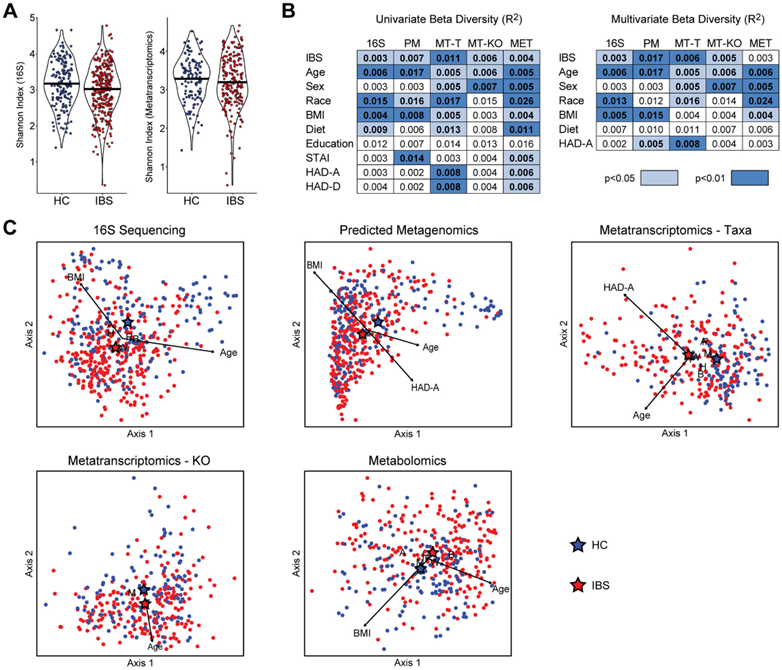
图1. 肠易激综合征与微生物组组成和功能的整体改变有关。
(a) 根据16S rRNA测序和宏录组学数据,显示了IBS受试者和健康对照(HC)粪便样本中微生物丰富度和均匀度的Shannon指数。(b) 通过 16S rRNA 测序数据的 Bray-Curtis 相异性、预测宏基因组学 (PM)、宏转录组学分类学 (MT-T) 和 KEGG 直系同源转录本注释 (MT-KO) 进行Beta多样性分析。(c) 进行基于距离的冗余分析(dbRDA)以可视化与IBS状态、年龄、性别、种族/民族、BMI、饮食类别和HAD-焦虑(HAD-A)相关的Beta多样性多样性的变化。
研究发现,IBS 的特征是Alistipes ihumii、Bacteroides dorei、Actinomyces odontolyticus和厚壁菌门的多个成员(例如Bartettii 肠杆菌和回肠Roumboutsia ilealis )丰度增加(图2A)。肠易激综合征显示Facealibacterium prausnitzii和Bacteroides thetaiotamicron丰度降低。在评估宏转录组的分类特征时,只有B. dorei与16S rRNA测序结果一致(图2B)。在肠易激综合征中转录本丰度增加的细菌包括:Eggerthella lenta、两类拟杆菌(B. dorei 和B. fluxus)、 Phascolarctobacterium succinatutens、Blautia hydrogenotrophica、Prevotella timonsensis、Clostridium hylemonae、Catonella morbi和一种未识别的放线菌。IBS显示Bilophila wadsworthia、Roseburia inulinivorans、双歧杆菌和两种拟杆菌(B.s plebeius和B.s barnesiae)的丰度降低。
通过细菌转录丰度[通过KEGG orthology (KO)进行注释]、利用系统发育最接近的参考基因组(预测宏基因组)从16S rRNA测序数据预测细菌基因丰度和代谢产物水平来评估微生物功能。在单变量分析中,IBS状态与所有三个数据集的β多样性变异显著相关(图1B)。协变量中,年龄、性别、种族、BMI、饮食类别和HAD-焦虑与宏转录组和代谢组显著相关;年龄、种族和BMI也与预测的宏基因组显著相关。在对这六个协变量进行调整后,IBS仍然与宏转录组和预测宏基因组显著相关,但不再与代谢组显著相关(图1B-C)。
差异丰度测试鉴定出16个细菌转录本在IBS中丰度增加,3个丰度降低(图2D)。在预测的宏基因组中,共有4个转录本出现了一致的变化,包括D-脯氨酸还原酶、1-2-二酰基甘油3-葡糖基转移酶和低聚果糖转运系统的两种组分(渗透酶和底物结合蛋白)的丰度增加。这表明,这些转录本水平的增加可归因于宏基因组中相应基因丰度的差异。宏转录组的其他显著变化包括丁酸激酶的丰度增加和甘露醇(一种可发酵的多元醇)摄取的磷酸转移酶丰度增加。
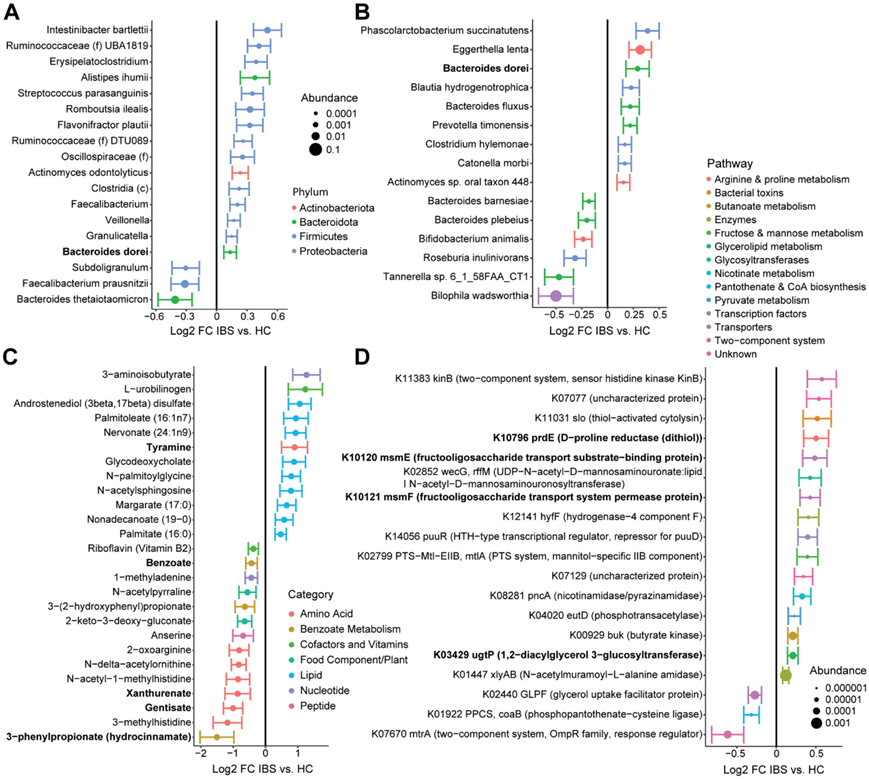
图2. IBS 的特点是细菌分类群、代谢物和转录本丰度发生改变,包括参与低聚果糖利用的基因。
(a-b) 16S rRNA 测序数据和宏转录组数据。(c) 显示了通过非靶向代谢组学检测到的差异富集的粪便代谢物。(d) 差异富集的细菌转录本。
02
组间分析揭示了不同数据集之间一致的代谢变化,将IBS与HC区分开来
他们进一步研究了三种组间关联。首先,评估了宏转录组(RNA)中微生物丰度与16S测序(DNA)中微生物丰度的比值,这些比值广泛代表了微生物的转录活性。与先前的报道一致,微生物的转录表现与它们在宏基因组中的丰度相比可能存在数量级的差异,从而区分出转录活跃的微生物和静止的微生物(图3A)。转录活性最高的属是韦永氏球菌属(HC组RNA/DNA中值为89,IBS组为114),转录活性最低的属是Faecalibacterium (HC组RNA/DNA中值为0.074,IBS组为0.087)。将IBS与HC进行比较,发现两组在属水平上具有相似的微生物转录活性模式(图3B),IBS与HC在分类上无统计学差异。
接下来,他们使用转录本丰度(RNA)与预测基因丰度(DNA)的比率计算基因标准化转录本丰度,用于差异基因表达分析。转录本与基因的比值范围很广,从小于0.1到大于1000不等(图3C)。虽然RNA/DNA比率在IBS和HC之间高度相关,但在调整协变量后,发现IBS和HC之间的182个KO存在差异调节(图3D)。
为了确定基因标准化转录本丰度之间一致的功能变化,他们进行了通路富集分析。柠檬酸循环、果糖和甘露糖代谢、原核生物的碳固定和氨基酰基tRNA生物合成的通路发生富集(图3E)。在这些通路中,21个差异转录本突出显示,这些转录本在IBS中都发生上调。这其中还涉及柠檬酸循环中的多个关键酶,包括丙酮酸羧化酶、磷酸烯醇式丙酮酸羧化酶、琥珀酸脱氢酶、延胡索酸水合酶和苹果酸脱氢酶。碳固定通路转录本包括乙酰-CoA合成酶、甲基丙二酰辅酶-CoA变位酶和甲基丙二酰-CoA向异构酶(它们参与短链脂肪酸丙酸的生成)。总的来说,这两种通路中的转录本几乎包含了碳水化合物发酵成丙酸琥珀酸通路中的所有酶(图3F)。
图3. 细菌分类和功能的组间比较表明,IBS 中参与碳水化合物发酵琥珀酸通路的转录本上调。
(a) 散点图描绘了具有两种数据类型的 322 名受试者的 16S rRNA 测序数据和宏转录组中属的中位相对丰度。 (b) IBS 受试者和 HC 宏转录组 (RNA) 与16S rRNA 测序数据 (DNA) 中属的丰度的中位数比率。(c) 预测宏基因组中基因丰度中值与转录本丰度中值相比的散点图。 (d) 绘制 IBS 受试者和 HC 的中位转录本/基因丰度比。(e) 鉴定出差异调节转录本显著富集的通路。 (f) 碳水化合物发酵的琥珀酸通路。
利用这些多组学特征,他们建立了随机森林分类器模型来区分 IBS和HC ,AUC 为 0.82,该模型在统计上显著优于使用单个数据集的微生物特征构建的分类器,其 AUC 为 0.67-0.70(图4A-B)。对这个分类器影响最大的三种微生物特征是代谢物:龙胆酸、氢化肉桂酸和酪胺。分类器中的其他十个特征包括与低聚果糖代谢相关的三个转录本(二磷酸依赖性磷酸果糖激酶、6-磷酸果糖激酶1、低聚果糖转运渗透酶蛋白)、柠檬酸循环中的三个转录本(丙酮酸羧化酶、苹果酸脱氢酶、富马酸水合酶)、谷氨酰胺酰-tRNA合成酶、D-脯氨酸还原酶、1,2-二酰基甘油3-葡萄糖基转移酶和甲基丙二酰-CoA差向异构酶。

图4. 与使用单个数据集的分类器相比,IBS 的多组学微生物组分类器显示出更高的准确性。
(a) 将根据五个数据集(红色)中每个数据集的差异丰富特征构建的随机森林分类器的 ROC 曲线与多组学分类器(蓝色)的 ROC 曲线进行比较。 (b) 显示多组学分类器中包含的特征的重要性分数,按特征类型着色。
03
细菌转录本和代谢物可区分肠易激综合征的肠习惯亚型
在确定了区分肠易激综合征和HC的强大微生物谱后,他们评估了肠易激综合征中微生物组成和功能与表型的关系。肠道习惯亚型与宏转录组学和代谢组学显著相关,但与微生物组组成或预测的宏基因组无关。在确定了区分 IBS 和 HC 的微生物特征后,他们评估了 IBS 内微生物组成和功能与表型的关系。排便习惯亚型与宏转录组学KO和代谢组学显著相关,但与微生物组组成或预测的宏基因组无关(图5A-B)。内脏敏感性 (VSI)、一般身体症状感知 (PILL) 和 IBS 特异性症状严重程度 (IBS-SSS) 与转录本、预测基因和代谢物的整体微生物组组成或功能均不显著相关。
为了进一步描述与肠易激综合征排便习惯亚型相关的微生物功能特征,他们重点比较了腹泻为主(IBS-D)和便秘为主(IBS-C)的亚型。差异丰度分析进一步证明IBS-D与IBS-C没有明确的分类学特征,因为16SrRNA测序数据显示只有2个分类群存在显著差异,宏转录组学数据显示只有4个分类群存在显著差异,两个数据集之间没有重叠。相比之下,宏转录组学功能评估显示54个差异转录本属于不同的通路,其中51个在IBS-D中增加。其中,许多与果糖和甘露糖代谢通路有关,包括L-二醇2-脱氢酶(多元醇脱氢酶),甘露糖特异性磷酸转移酶系统的三个组成部分,以及L-岩藻糖 / D-阿拉伯糖异构酶(产生核酮糖)。参与D-谷氨酸合成(谷氨酸合成酶和谷氨酸消旋酶)和乙醇胺利用的转录本也有富集。这些都没有与差异预测的基因丰度重叠。
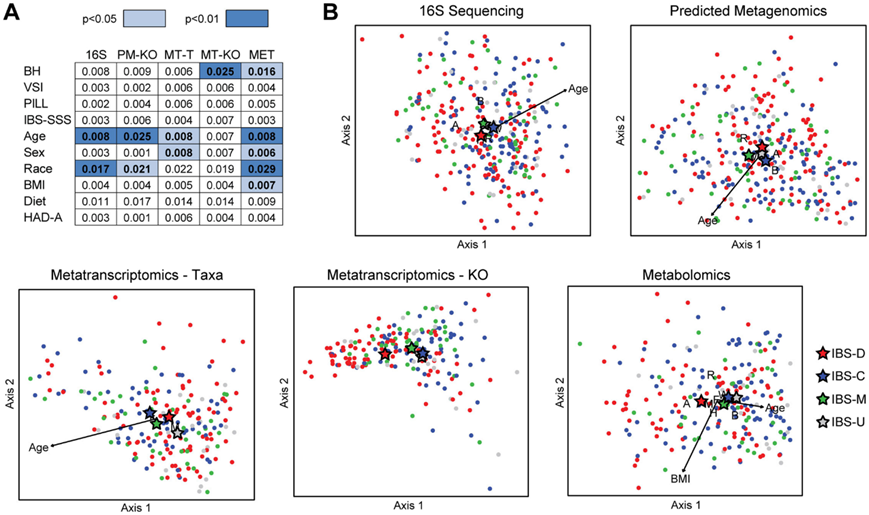
图5. 根据宏转录组学和代谢组学研究,IBS 肠道习惯 (BH) 亚型具有不同的功能特征。
(a) 使用多变量 PERMONOVA 模型评估 IBS 内表型的关联,包括 BH 亚型、内脏敏感性 (VSI)、一般身体症状感知 (PILL) 和 IBS 严重程度 (IBS-SSS) 与五个数据集的关系。 (b) 5个数据集的DbRDA图显示了与BH亚型和显著分类或连续协变量相关的Beta多样性差异。
04
IBS-D 的特征,并区分 IBS-D 和 IBS-C
他们进行基因标准化转录丰度的组间分析,以评估IBS-D与IBS-C的转录调节模式。IBS-D与140个转录本RNA/DNA比率的显著变化相关,其中128个在IBS-D中上调(图6A-B)。通路分析显示,10条通路富集了差异富集的转录本,包括果糖和甘露糖代谢、丙酸代谢、氨基酰基- tRNA生物合成、萜类生物合成以及戊糖和葡萄糖酸盐相互转化。最显著上调的转录本涉及丙酸代谢,包括2-甲基柠檬酸合成酶、丙二醇脱氢酶的三个亚基和甘油脱氢酶。
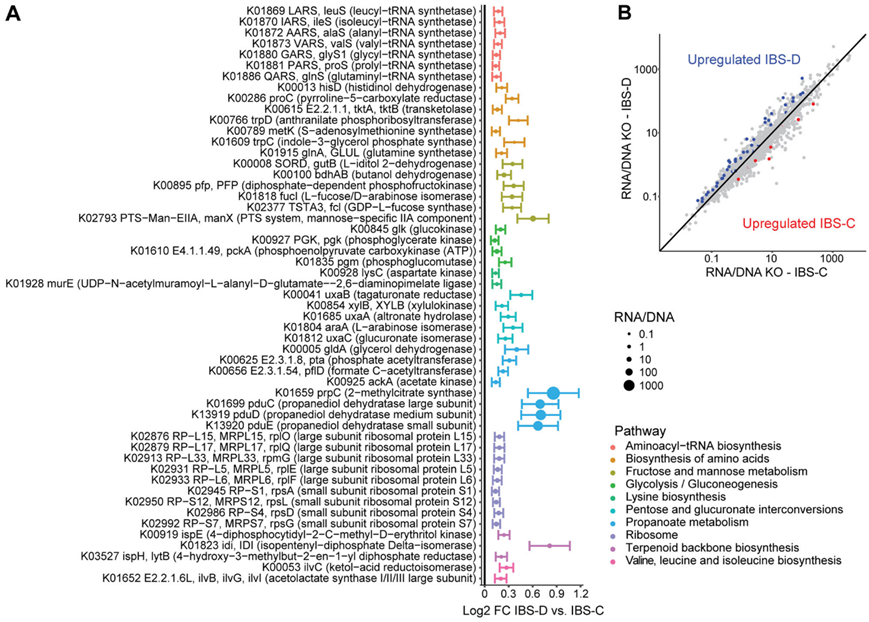
图6. IBS-D 的特征是相对于 IBS-C 涉及丙酸代谢、萜类生物合成、果糖和甘露糖代谢以及糖酵解的基因转录上调。
(a) 鉴定出在 IBS-D 与 IBS-C 中差异调节的转录本显著富集的通路。(b) 鉴定出在 IBS-D 与 IBS-C 中差异调节的转录本显著富集的通路。
然后,他们评估了差异转录本、代谢物和多组学特征(基因标准化转录丰度和与微生物代谢潜力显著相关的差异代谢物)鉴别IBS-D和IBS-C的能力。多组学分类器的性能最高,AUC为0.86,显著大于预测的宏基因组的AUC为0.65(图7A)。由宏转录组学 KO 和代谢物构建的分类器各自的 AUC 为 0.79,与多组学分类器没有显著差异。宏转录组学分类器中的转录本包括乙醇胺利用蛋白、丙二醇脱水酶、钠/脯氨酸同向转运蛋白和N-乙酰氨基葡萄糖磷酸转移酶(图7B)。代谢组学分类器包含氨基酸发酵的所有三种不同产物(尸胺、N-乙酰尸胺、N-乙酰腐胺)、苹果酸、脂肪酸(肉豆蔻酸和2-棕榈酰甘油)和双半乳糖醛酸(果胶分解的产物)(图7C)。有助于多组学分类的特征包括果糖和甘露糖代谢(二磷酸依赖性磷酸果糖激酶,GDP-L-聚焦合酶)、萜类生物合成(异戊烯-二磷酸δ -异构酶,4-羟基-3-甲基丁基-2-烯-1-基二磷酸还原酶,4-二磷酸胞苷基-2-c-甲基-d-赤糖醇激酶)、戊糖和葡糖醛酸相互转化(L-阿拉伯糖异构酶)和丙酸代谢(2-甲基柠檬酸合成酶、甘油脱氢酶)(图7D)。
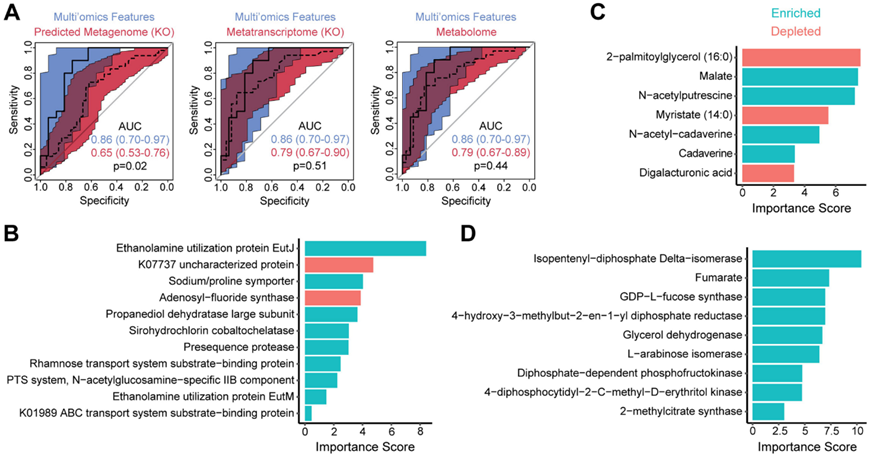
图7. 代谢物、转录本和转录本/基因比率可以高精度地区分 IBS-D 和 IBS-C。
(a) 将由差异丰富的预测基因、转录本和代谢物(红色)构建的随机森林分类器的 ROC 曲线与多组学分类器(蓝色)的 ROC 曲线进行比较。 (b-d) 图中显示了转录组学、代谢组学和多组学分类器中包含的特征的重要性评分,条形颜色表示IBS-D中富集或缺失的特征。
+ + + + + + + + + + +
结 论
本项研究通过 16S rRNA 测序和宏转录组学,IBS 与微生物组组成的整体变化相关;通过预测的宏基因组学、宏转录组学和代谢组学,IBS 与微生物组功能的整体变化相关。在调整年龄、性别、种族/民族、BMI、饮食等参数后,IBS 与细菌类群(如Bacteroides dorei)的差异丰度相关。代谢物包括酪胺增加和龙胆酸和氢化肉桂酸减少;以及与低聚果糖和多元醇利用相关的转录本。IBS 进一步显示参与果糖和葡聚糖代谢以及碳水化合物发酵的琥珀酸通路的酶的转录上调。IBS 多组学分类器的准确度 (AUC 0.82) 明显高于使用单个数据集的分类器。与便秘为主的 IBS 相比,腹泻为主的 IBS (IBS-D) 表现出宏转录组和代谢组的变化,包括胆汁酸、多胺、琥珀酸通路中间体(苹果酸、富马酸)以及参与果糖、甘露糖和多元醇代谢的转录本增加。结合代谢物和基因标准化转录本的分类器以高精度区分 IBS-D 和 IBS-C(AUC 0.86)。
+ + + + +



