

 English
English文献解读|Cell Rep Med(14.3):对免疫治疗耐药的微卫星不稳定性高胃肠癌患者肠道微生物生态系统的多组学研究
✦ +
+
论文ID
原名:Multi-omics of the gut microbial ecosystem in patients with microsatellite-instability-high gastrointestinal cancer resistant to immunotherapy
译名:对免疫治疗耐药的微卫星不稳定性高胃肠癌患者肠道微生物生态系统的多组学研究
期刊:Cell Reports Medicine
影响因子:14.3
发表时间:2024.01.08
DOI号:10.1016/j.xcrm.2023.101355
背 景
错配修复缺陷 (dMMR) 亚型占胃肠道 (GI) 癌症患者的 15%–22%,其中约 5% 是转移性/复发性胃肠道癌症。dMMR亚型患者无法识别和修复某些自发突变,导致相当高的肿瘤突变负担和高微卫星不稳定性(MSI-H)状态,因此更有可能受益于抗PD-1/ PD-L1(靶向程序性细胞死亡蛋白 1 [PD-1] 或其配体 PD-L1 的抗体)免疫疗法。然而,MSI-H/dMMR 胃肠道癌症患者对免疫治疗表现出高度异质性的反应,大约 30% MSI-H 患者对免疫治疗表现出原发性耐药。因此,了解影响疗效异质性的其他因素对于增强 dMMR/MSI-H 患者的免疫治疗反应非常重要。
实验设计
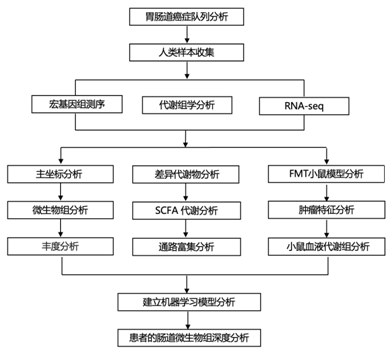
结 果
01

晚期 MSI-H/dMMR 胃肠道癌症队列分析
2018年2月至2020年10月,北京肿瘤医院治疗了98例MSI-H/dMMR晚期胃肠癌患者,其中胃癌(GC)患者30例,结直肠癌(CRC)患者68例。如果患者在治疗开始后获得持续至少 6 个月的客观缓解(完全缓解 [CR]/部分缓解 [PR]/疾病稳定 [SD]),研究者团队将其定义为缓解者 (R)否则定义为非缓解患者(NR)(治疗开始后 6 个月内观察到的进展性疾病 [PD])。基线定义为治疗前或免疫治疗后不超过3周。他们在基线和治疗期间共收集了290份粪便样本(图1A-B)。同时,他们还收集了一部分患者(15例GC和55例CRC)的基线血液样本(血浆),用于代谢组学分析和细胞因子/趋化因子分析。
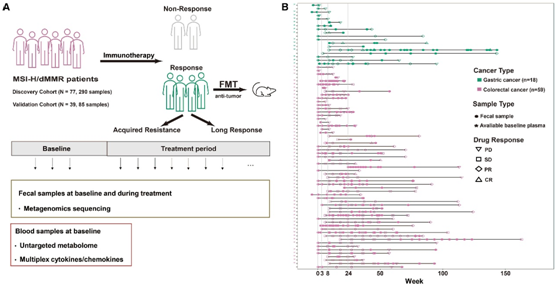
图1. 研究设计和临床样本采集。
(A)招募了 18 名患有 MSI-H/dMMR 的胃癌 (GC) 患者和 59 名结直肠癌 (CRC) 患者进行分析。(B) 总共收集了 290 份粪便样本和 70 份血浆样本进行分析。
02
对免疫治疗具有原发耐药性的肠道微生物组特征
为了探索与免疫治疗原发性耐药相关的整体特征,他们对基线样本进行了多组学分析。首先分析了从粪便样本中收集的宏基因组数据。整体肠道微生物结构的 Beta 多样性(主坐标分析,PCoA)显示 R和 NR 患者之间没有显著差异(图2A)。为了找到MSI-H患者的共同特征并避免癌症类型的影响,他们进行了组合分析,并通过MaAsLin2(微生物组多变量与线性模型的关联)分析。他们探索了 R 和 NR 患者之间显著改变的物种,发现Bacteroides caccae、Porphyromonadaceae、Parabacteroides、Acidaminococcaceae、Alistipes finegoldii、Paraprevotella clara、Bacteroides massiliensis和Alistipes putredinis在R中富集,而Veillonella parvula、Veillonella atypica、Peptostreptococcaceae、Streptococcus thermophilus和Micrococcaceae在NR中富集(图2B)。包括纵向样本在内的时间序列分析显示,R和NR患者之间存在11种差异,其中,NR样本中V. parvula和V. atypica的丰度一致较高,而R样本中Coprobacter、A. putredinis、B. caccae和B. clarus的丰度一致较高。
然后,他们使用液相色谱-质谱法 (LC-MS) 对基线血浆样本进行了整体代谢组学分析。R和 NR 患者之间存在143 种代谢物的差异(图2C)。其中,两种代谢物值得注意,第一类是精氨酸代谢相关的代谢物,如精氨酸、鸟氨酸、谷氨酰胺、不对称二甲基精氨酸、天冬酰胺、丝氨酸、瓜氨酸和脯氨酸。他们还分析了精氨酸和肠道微生物之间的关系,发现了几种与精氨酸数量显著相关的微生物,包括Paraprevotella clara和V. atypica,这些微生物是R和NR之间的差异特征。另一类代谢物与短链脂肪酸代谢相关,如4-氨基丁酸、癸酸、2-酮己酸、4-乙酰氨基丁酸和3-羟基丁酸。他们还对短链脂肪酸 (SCFA)进行了靶向代谢组学研究,发现R样本中丁酸盐、己酸盐和异丁酸盐显著富集(图2D)。R组和NR组之间的途径富集分析也表明,与SCFA代谢相关的丙酸代谢、精氨酸和脯氨酸代谢具有显著意义(图2E)。
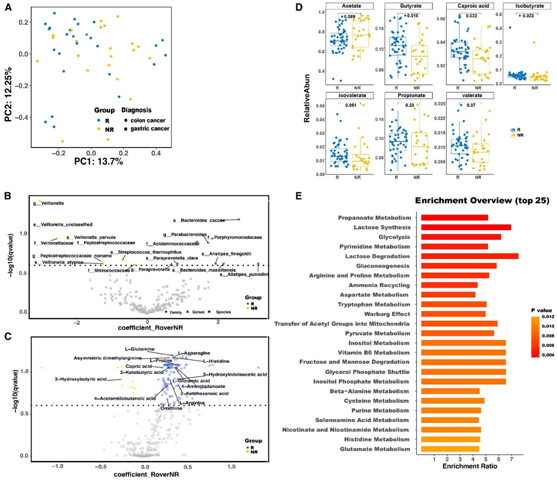
图2. 与药物反应相关的肠道微生物组组成、功能途径和血液代谢物。
(A) 使用 Bray-Curtis 相异性按响应和诊断排列的粪便样本的 PCoA 图。(B) 基于 MaasLin2 结果的 R和 NR患者之间基线处丰富度差异的类群火山图。(C) 基于 MaasLin2 结果的 R和 NR患者之间代谢组丰度差异的火山图。(D) 基于 Wilcoxon 检验,R和 NR之间不同短链脂肪酸相对丰度的差异。(E) 基于非靶向代谢组数据,R 富集的前 25 个功能途径。
03
粪便微生物群移植 (FMT)对小鼠的临床益处
为了验证肠道微生物组在介导患者免疫治疗反应中的作用,他们将6例患者(3例R, 3例NR)的粪便样本移植到经广谱抗生素预处理的MC38小鼠模型中。在评估FMT联合抗PD1治疗的潜在协同效应时(图3A),他们观察到,与接受NR患者粪便(NR组)或单独接受抗PD1治疗(阴性对照[NC]组)的小鼠相比,移植R患者粪便的小鼠(R组)的肿瘤体积显著缩小(图3B)。利用多重免疫化学(mIHC)评估肿瘤微环境(TME)中的T细胞群,他们观察到,在接受R粪便的小鼠中,辅助性T细胞(CD3+CD4+CD8−)、细胞毒性T细胞(CD3+CD4−CD8+)和调节性T (Treg)细胞(CD4+CD8−FoxP3+)的比例增加(图3C-D)。这些结果表明,R患者粪便样品的FMT可以改变肿瘤微环境(TME)中的免疫细胞景观,并增强免疫治疗。
他们进一步进行了多组学分析,以分析R组和NR组之间的差异,包括小鼠血液样本的代谢组学分析和小鼠肿瘤组织的转录组分析(RNA-seq)。共鉴定出97种差异代谢物,包括精氨酸,此前在R患者中发现其含量较高(图3E)。通路富集分析也确定了精氨酸生物合成的显著性(图3F),其中R组参与尿素循环的四个基因(一氧化氮合酶2 [NOS2]/gm5424/arg1/arg2)的表达上调。这些结果表明,活跃的氨代谢和精氨酸合成可能影响免疫检查点抑制剂(ICI)反应。
此外,他们分析了血液代谢物与肿瘤免疫细胞群的关系,发现与精氨酸代谢相关的代谢物不对称二甲基精氨酸与Treg细胞显著相关,提示精氨酸代谢在调节免疫功能中的作用(图3G)。
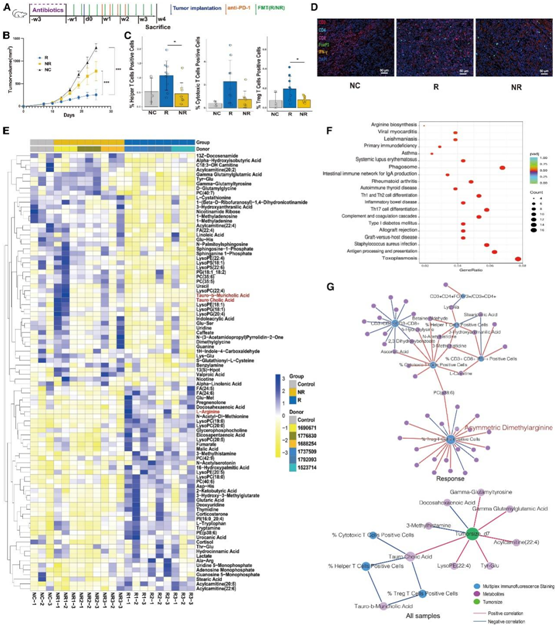
图3. 使用Rs粪便进行粪便微生物群移植(FMT)后小鼠的抗肿瘤效果增强。
(A) 实验设计。(B) 肿瘤生长曲线。(C) 肿瘤微环境中的辅助 T 细胞 (CD3+CD4+CD8−)、细胞毒性 T 细胞 (CD3+CD4−CD8+) 和 Treg (CD4+CD8−FoxP3+) T 细胞群,通过 mIHC 进行定量。(D) 肿瘤组织样本的 mIHC 代表性图像。(E) 热图显示从血液代谢组中鉴定出的差异代谢物。(F) 点图显示基于小鼠肿瘤基因表达数据的显著富集的 KEGG 通路。(G) 肿瘤微环境中肿瘤大小、血液代谢物和 T 细胞群的网络分析。
04
基于肠道微生物组的机器学习模型很好地预测了免疫治疗的疗效
他们试图找出在ICI治疗前预测反应概率是否可行,并利用肠道微生物组数据确定与临床结果相关的生物标志物。为了实现这一目标,他们用微生物物种数据构建了LightGBM机器学习模型(图4A)。该模型使用具有基线样本的患者数据进行训练,并进行10倍交叉验证以评估模型的性能。为了评估模型的稳健性,他们收集了另一个独立的验证队列,包括31个基线粪便样本。该训练模型在使用微生物种类数据预测ICI治疗效果方面表现出色。具体来说,他们在训练数据集中实现了0.9的曲线下面积(AUC),在测试集中实现了0.88,在独立验证集中实现了0.79(图4A)。通过分析特征对模型预测反应状态的作用,他们发现B. caccae、V. parvula、V. atypica和Clostridiales是ICI反应的前4个指示性特征(图4B)。值得注意的是,前三者是有显著差异的。这些结果强调了肠道微生物组作为一种预测因素的潜力,可以识别可能从免疫治疗中受益的患者,所选择的微生物物种可能是预测ICI治疗反应的潜在生物标志物。

图4. 基于微生物生物特征的机器学习模型。
(A) 基于物种的机器学习模型的受试者工作特征 (ROC) 曲线 (AUC = 0.76)。(B) 上述三个机器学习模型中以SHAP值表示的特征。
05
获得性免疫治疗耐药患者的生物特征
在临床实践中,相当多的患者对免疫治疗产生了获得性耐药(AR),定义为患者在客观反应或SD延长6个月以上后,对ICI仍无反应。另一方面,长R(LR)定义为在分析时具有超过1年的持续获益且无疾病进展。因此,为了了解肠道微生物组在AR过程中的作用,他们进一步将R分为AR组和LR组。基线多组学分析确定了AR和LR样本之间的一些差异特征(微生物、代谢物或细胞因子/趋化因子),包括AR组中白细胞介素-5 (IL-5)、氢肉桂酸、2- 16烯醛和胆绿素的显著增加,以及LR组中水杨酸和糖醛酸的显著增加。为了系统地了解肠道微生物组在AR中的作用,他们进一步分析了患者获得ICI耐药性前后的肠道微生物组,共鉴定出18种与耐药相关的菌株(图5A)。值得注意的是,两种Alistipes (A. onderdonkii和A. putredinis)在AR样品中含量较低,但在初级耐药组(NR)中含量也有所下降,这表明它们在ICI耐药中可能发挥作用。在耐药过程中,Ruminococcus bromii、Bifidobacterium adolescentis、Bifidobacterium pseudocatenulatum等菌种明显减少,并且在LR患者中也发现它们长期增加(图5B)。
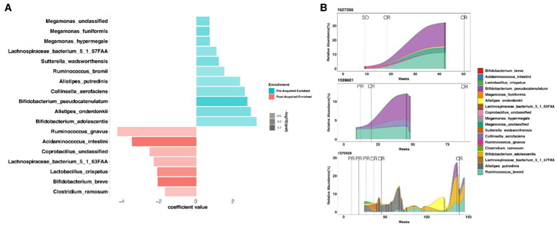
图5. 与 AR 相关的肠道微生物组。
(A) 在耐药性进展过程中鉴定出的差异微生物。(B) 代表性 LR 患者的微生物动态,重点关注 AR 进展过程中发现的差异微生物。
+ + + + + + + + + + +
结 论
本项研究使用多组学方法,在基线和治疗期间分析 MSI-H/dMMR 胃肠癌患者的肠道微生物组、血液代谢组和细胞因子/趋化因子。他们确定了许多与免疫治疗的原发性耐药性高度相关的微生物(例如卟啉单胞菌科)和代谢物(例如精氨酸)。使用独立验证队列和小鼠模型来进一步验证。还建立了主要阻力的预测机器学习模型,并在外部验证集上实现了 0.79 的准确度。此外,还确定了几种微生物在获得性耐药过程中逐渐发生变化。总之,本项研究证明了肠道微生物组在耐药性中的重要作用,这可以作为未来的预防性诊断工具和治疗靶点。
+ + + + +



