

 English
English文献解读|Genome Med(12.3):结直肠癌微生物组通过影响甲基供体代谢来编程宿主细胞的 DNA 甲基化
✦ +
+
论文ID
原名:Colorectal cancer microbiome programs DNA methylation of host cells by affecting methyl donor metabolism
译名:结直肠癌微生物组通过影响甲基供体代谢来编程宿主细胞的 DNA 甲基化
期刊:Genome Medicine
影响因子:12.3
发表时间:2024.06.15
DOI号:10.1186/s13073-024-01344-1
背 景
结直肠癌 (CRC) 源于宿主与环境之间复杂的相互作用,其中包括肠道和组织微生物群。据推测,肠道微生物群的表观遗传调控是共生微生物动态影响肠道生物学的基本界面。本研究旨在探索 CRC 中肠道和组织微生物群与宿主 DNA 甲基化之间的相互作用。
实验设计
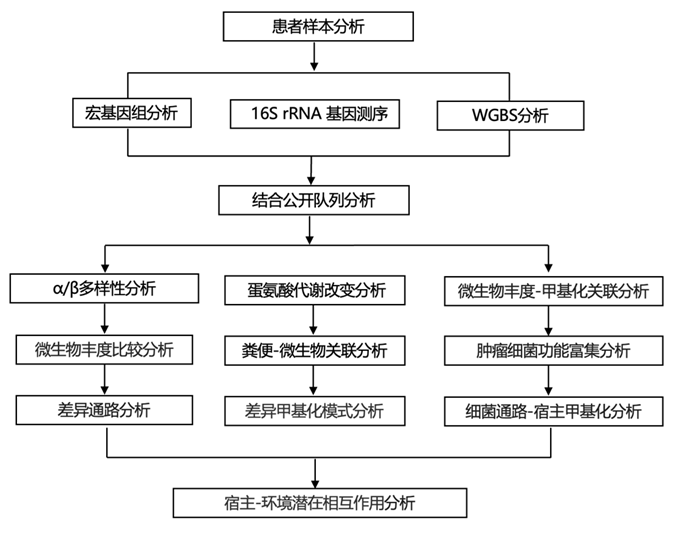
结 果
01

CRC 肠道微生物组中细菌和细菌甲基供体相关功能的改变
研究团队对 18 名 CRC 患者和 18 名健康对照 (HC) 的粪便样本进行了宏基因组测序(图 S1A)。通过丰富度、Shannon、辛普森 (Simpson)、Pielou、Chao 和 ACE 指数评估了表示群落丰富度、多样性和均匀度的 Alpha 多样性指数。在 CRC 和 HC 的比较中,未观察到 α 多样性的统计学显著变化 (图 S2)。他们进行了偏最小二乘判别分析 (PLS-DA) 分析以评估 β 多样性。PLS-DA 分析表明,CRC 患者的整体粪便微生物组群落与 HC 不同(图 1 A)。在HC和CRC患者之间的相对物种丰度的比较中,他们发现在CRC患者的粪便样本中,一些先前报道的与CRC相关的物种富集,如具核梭杆菌和多形拟杆菌(图1B)。此外,对与 CRC 相关的微生物功能的研究确定了 35 种符合标准的细菌通路(图1C),突出观察到了多种与蛋氨酸代谢相关的通路(图 1 C)。例如,COBALSYN − PWY(从钴胺素 I 中回收的腺苷钴胺)在 HC 中表现出富集。腺苷钴胺素,也称为辅酶 B12,可作为辅酶,在蛋氨酸合成过程中实现甲基的转移。尽管如此,与四氢叶酸生物合成相关的通路,包括四氢叶酸生物合成通路和四氢叶酸生物合成与回收通路,在CRC 样本中表现出明显的过表达。四氢叶酸及其衍生物统称为叶酸,是单碳代谢中不可或缺的辅助因子。这些分子实体在运输和补充 C1 单元方面发挥着关键作用,这对于蛋氨酸和其他各种代谢物的合成至关重要。
为了进一步验证蛋氨酸代谢失调在结直肠癌中的发现,他们对来自10个公共结直肠癌队列的粪便宏基因组数据进行了Meta分析(图S1B)。他们观察到蛋氨酸代谢相关通路发生了显著改变。特别是,S-腺苷-L-蛋氨酸循环 I 通路(其中 S-腺苷-L-同型半胱氨酸再循环回 SAM)在各个队列的 CRC 粪便样本中减少(图 1 D-E)。这些发现进一步证明了,与 HC 相比 CRC 中甲基供体相关通路失调。
图S1. 研究概述。
(A)粪便样本的宏基因组测序。(B) CRC和健康对照的粪便微生物组的Meta分析。(C)肿瘤和匹配的癌旁组织的16s rRNA测序和WGBS测序。(D)肿瘤和AN组织的组织微生物组的Meta分析。(E)不同水平粪便和组织微生物组的比较。(F-G)队列的甲基化-微生物关联分析。
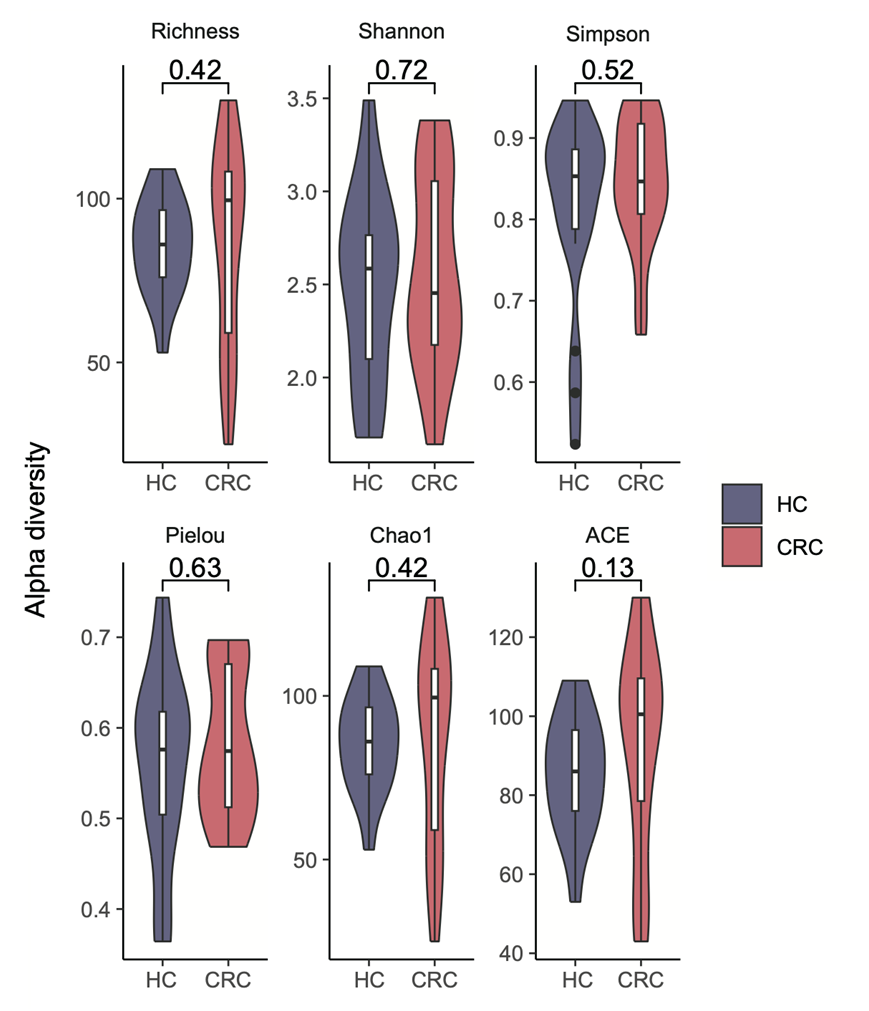
图S2. 来自结直肠癌患者和健康对照的粪便微生物组的α多样性。
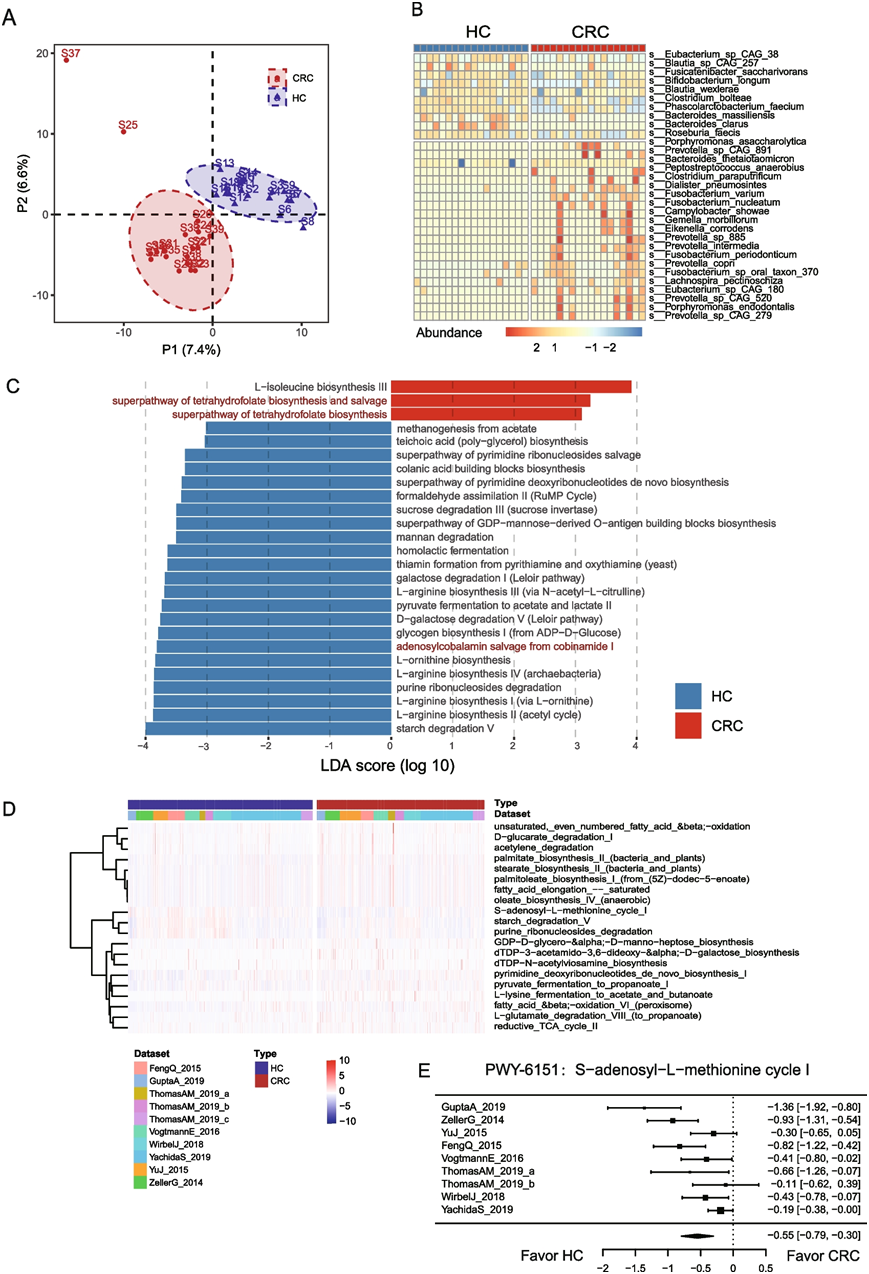
图1. CRC 患者和健康对照者的粪便微生物组。
(a) 通过 PLS-DA 分析评估的β多样性。(b) CRC和健康对照 (HC) 之间的差异细菌种类。(c) CRC和 HC 之间的差异细菌通路。(d)结合 10 个公共队列的 CRC 和 HC 的粪便微生物组的Meta分析确定的前 20 条差异通路。(e) S-腺苷-L-蛋氨酸循环 I 通路的森林图。
02
肿瘤组织微生物组中蛋氨酸代谢改变
为了直接探索肿瘤组织中与微生物组有关的微环境,他们使用 16S rRNA 基因扩增子测序对 24 对肿瘤和邻近正常 (AN) 生物种进行了微生物组分析(图 S1C)。配对分析显示,CRC 患者的肿瘤和 AN 组织之间的微生物结构和组成存在差异(图2 A-C)。虽然肿瘤和 AN 组织之间的 alpha 多样性仍然相当,但在 β 多样性以及多个分类学水平的微生物丰度方面观察到了显著差异(图2 A-C)。与CRC相关的细菌,如Parabacteroides、Alistipes和Ruminococcus,在肿瘤组织中显著富集。此外,新发现的CRC 病原体Clostridium hathewayi在肿瘤组织中有显著的过度表达。为了进一步研究 CRC 中改变的微生物群的功能影响,他们利用 PICRUSt2 预测微生物组的 MetaCyc 通路和酶功能。具体而言,他们发现与蛋氨酸代谢相关的功能和通路显著富集。例如,“腺苷甲硫氨酸脱羧酶”和与L-甲硫氨酸生物合成通路在AN组织中显著富集(图2D-E)。腺苷蛋氨酸脱羧酶催化SAM转化为S-腺苷蛋氨酸,在蛋氨酸回收循环中起关键作用。与肠道菌群分析一致,这些结果进一步强调了蛋氨酸代谢在维持结肠组织正常DNA甲基化模式中的潜在参与。
接下来,他们将本研究的数据与来自CRC和匹配AN组织的另外四个16S rRNA测序数据集结合起来进行了Meta分析(图S1D)。基于预测的MetaCyc通路和酶委员会(Enzyme Commission, EC)分类进行分析。他们注意到L-蛋氨酸生物合成III通路在肿瘤样本中富集,而蛋氨酸合成酶在邻近正常样本中富集(图2F-G)。这些结果强调了肿瘤和邻近正常组织之间蛋氨酸代谢的实质性改变。
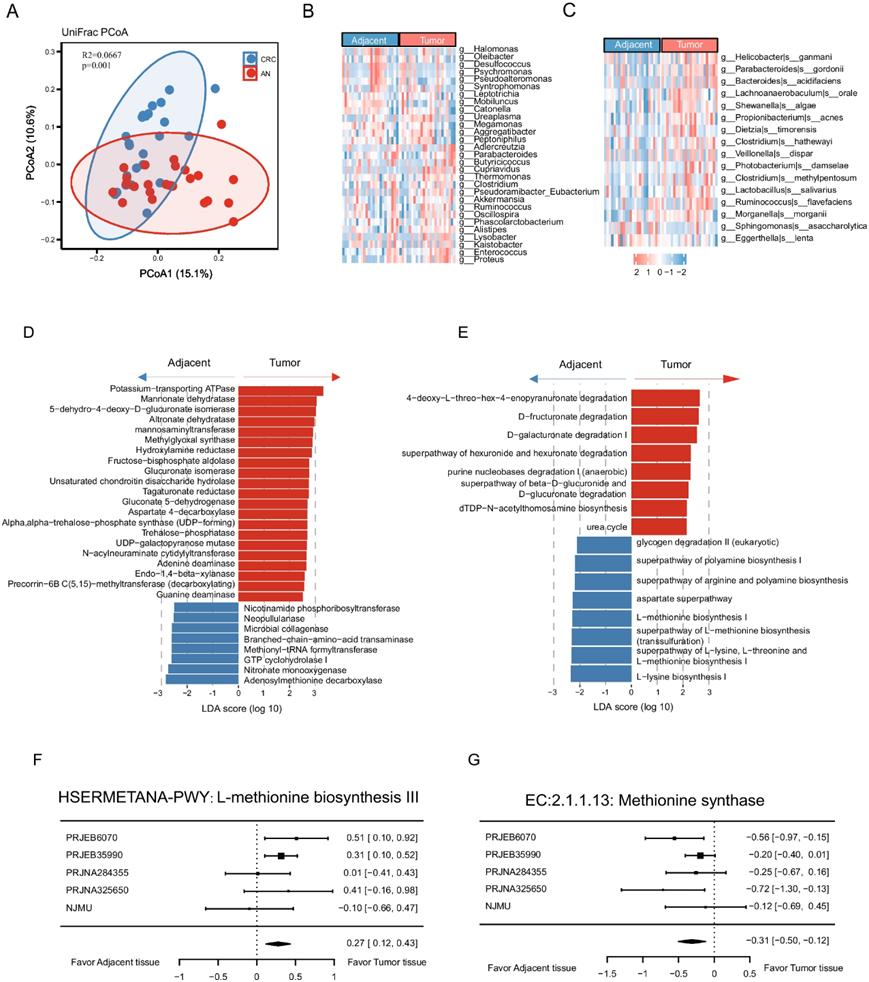
图2. 肿瘤和邻近正常组织中的微生物组概览。
(a) 通过 PCoA 分析评估的β多样性。(b-c) CRC和 AN 组织在属和种水平上的差异分类群。 (d-e) 肿瘤和 AN 组织之间的差异微生物酶功能和 MetaCyc 通路。 (f-g) L-蛋氨酸生物合成III通路相对丰度与蛋氨酸合酶功能之间关联的森林图。
03
CRC 中粪便和组织微生物组之间的关联
微生物组研究中的一个关键争论涉及粪便中肠道微生物组成与组织中肠道微生物组成之间的相关性。他们通过检测CRC患者的粪便微生物组与来自肿瘤和 AN 组织的组织微生物组之间的关联来研究这种关系(图 S1E)。在属的水平上,粪便微生物组的丰富度与肿瘤或 AN 组织的组织微生物组的丰富度没有任何明显的关联(图 3 A)。然而,衡量微生物组丰富度和均匀度的 Shannon 指数在组织样本中明显更高(图 3 B)。β 多样性分析显示,粪便样本的微生物组与组织样本的微生物组不同(图 3 C),粪便微生物组与肿瘤微生物组的距离略高于与 AN 微生物组的距离(图 3 D)。
粪便和组织微生物组之间的分类群相关性分析揭示了一些显著的关联。例如,艾肯菌(Eikenella)的丰度在粪便和组织之间显著相关。艾肯菌是口腔、肠道和生殖道的常见生物,也是粘膜微生物群的一部分,它是一种导致各种感染的机会性病原体。
接下来,他们进一步评估了粪便和组织微生物组之间微生物功能的一致性。值得注意的是,观察到粪便微生物组和肿瘤组织微生物组之间 23 条通路的丰度呈正相关。同样,12 条通路显示出粪便微生物组和 AN 组织微生物组之间的正相关。这些关联包括 13 条肿瘤组织特有的通路、12 条肿瘤和 AN 组织之间共有的通路以及 2 条肿瘤组织独有的通路(图 3 E)。这一有趣的发现表明,粪便细菌的功能属性可能部分反映了肿瘤组织的功能属性。例如,粪便样本中的 L-谷氨酸降解 VIII 通路与肿瘤组织中的相同通路表现出很强的相关性,而在 AN 组织中并未观察到这种相关性(图 3 F)。值得注意的是,使用公开的粪便微生物组数据进行的Meta-分析表明,肿瘤样本中 L-谷氨酸降解 VIII 通路显著增加(图 1 D)。这一系列证据表明,粪便微生物组内的特定功能可能确实反映了 CRC 肿瘤组织内的功能。
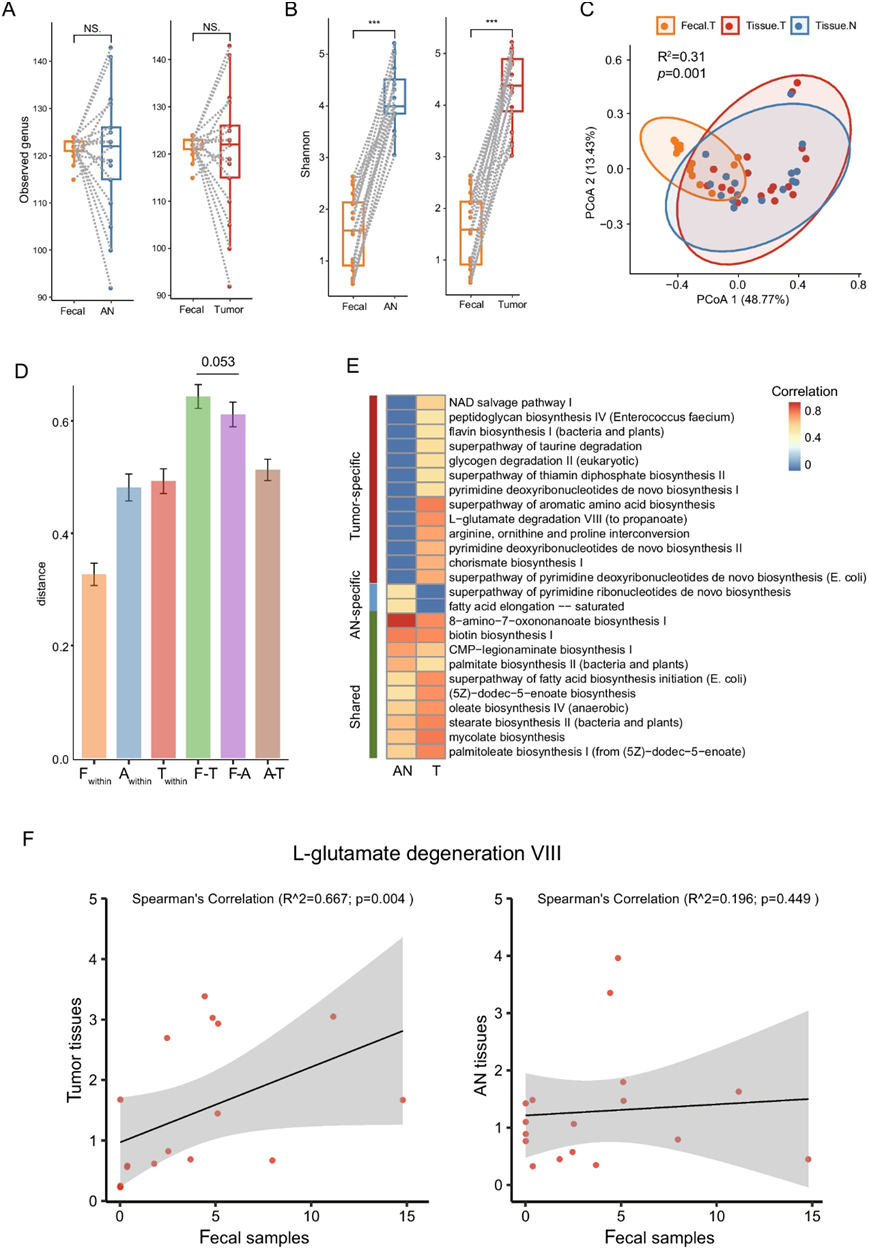
图3. CRC 中粪便和组织中微生物组之间的关系。
(a-b) CRC 中粪便和组织中微生物组之间的关系。(c-d) 通过Bray-Curtis 距离的 PCoA 计算粪便、肿瘤和 AN 组织微生物组之间的 Beta 多样性。 (e) 粪便微生物组与肿瘤或 AN 组织微生物组之间存在正相关的细菌通路。 (f)肿瘤特异性相关性的示例通路。
04
CRC 肿瘤和邻近正常组织中不同的 DNA 甲基化谱
使用全基因组亚硫酸盐测序 (WGBS) 对 13 个 CRC 组织活检及其对应的AN进行了全基因组 DNA 甲基化分析(图 S1C)。与匹配的 AN 组织相比,CRC 组织表现出明显较低的整体甲基化水平(图 4 A)。主成分分析 (PCA) 显示肿瘤和 AN 组织之间的甲基化谱存在明显差异(图 4 B)。通过使用来自 TCGA-CRC 队列的 45 组配对肿瘤和 AN 组织进行额外分析,进一步验证了这些发现(图4C-D)。
为了全面研究全基因组差异甲基化模式,他们确定了差异甲基化位点 (DML) 和区域 (DMR)。根据 CRC 和 AN 组织之间的绝对平均甲基化差异 (delta) > 0.2,将这些区域归类为高甲基化或低甲基化,代表甲基化水平变化 20%。在 CRC 中共鉴定出 210171 个低甲基化区域和 1210 个高甲基化区域。值得注意的是,启动子区域高度低甲基化(图 4 E-F)。此外,功能富集分析显示差异甲基化基因在 G 蛋白偶联受体信号通路和离子转运功能中富集(图 4 G),这与基于 TCGA-CRC 队列数据的分析一致
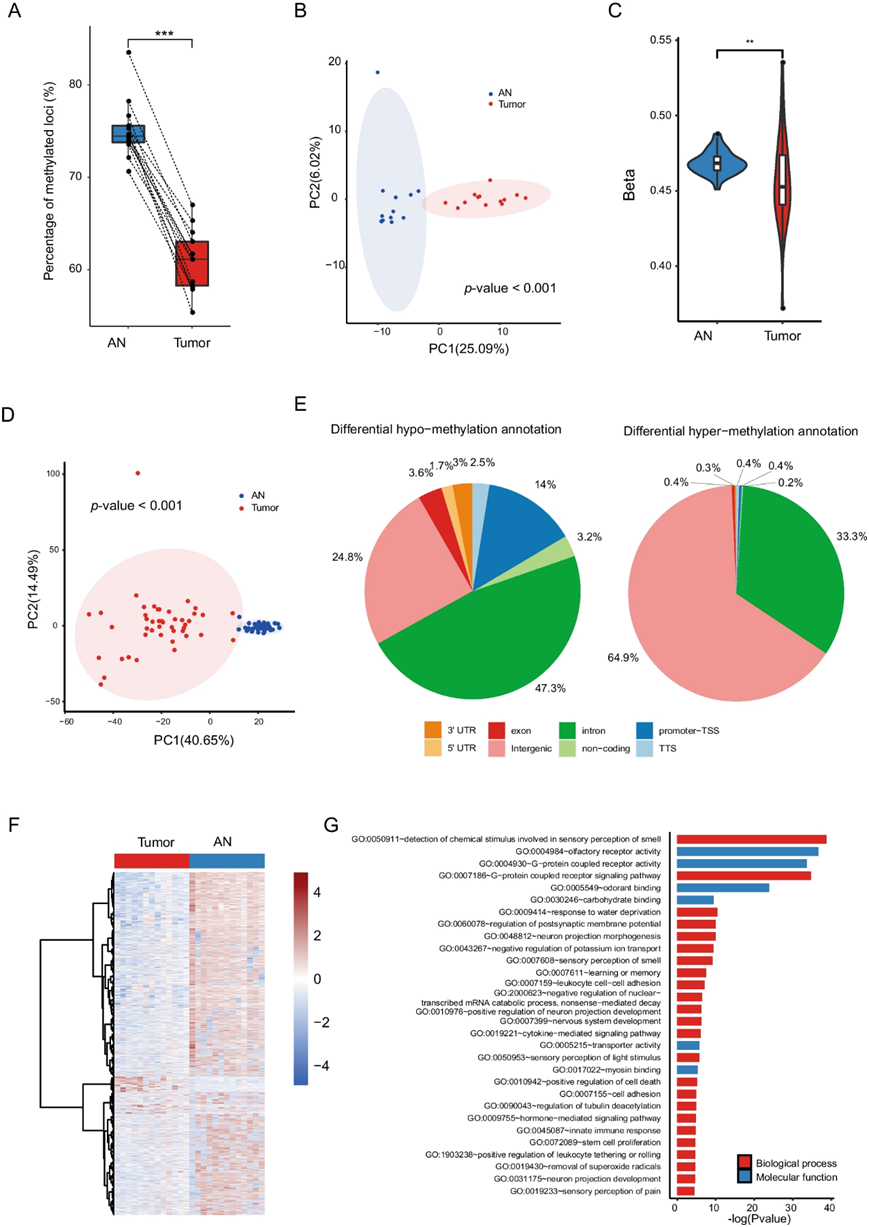
图4. 肿瘤和邻近正常 (AN) 组织中的 DNA 甲基化概况。
(a) CRC组织和其匹配的邻近正常组织的整体甲基化水平的成对比较。(b) PCA图区分了肿瘤和 AN 组织。(c) TCGA CRC 肿瘤和正常生物种的平均甲基化水平。(d) PCA 图区分了 TCGA-CRC 队列中的肿瘤和 AN 组织。(e) 基因组区域中显著低甲基化和高甲基化区域的分布。(f) 启动子区域内差异甲基化位点的热图。(g) 差异甲基化基因的功能富集。
05
肿瘤组织中的微生物启动子甲基化关联发生扰乱
为了深入了解结直肠癌 (CRC) 组织中微生物群与 DNA 甲基化之间的相互作用,他们研究了微生物丰度与宿主甲基化水平之间的关联。首先,进行了一项综合分析,将属和种水平的细菌分类群丰度与宿主肿瘤AN组织中的总体甲基化水平相关联(图 5A),该分析的结果清楚地表明,与整体 CpG 甲基化水平相关的细菌属和种的丰度在肿瘤和 AN 组织之间表现出明显差异。这一有趣的发现表明,在结直肠癌的背景下,微生物组成及其对 DNA 甲基化的影响在肿瘤组织和 AN 组织之间可能存在显著差异。
随后,他们探索了单个宿主 CpG 基因座与细菌类群之间的关联,采用lasso惩罚回归模型来识别特定的微生物类群,这些类群的丰度与整个基因组中 CpG 基因座的甲基化水平相关(图 S1F)。这些模型以基因座方式拟合,以每个宿主 CpG 基因座的甲基化水平作为因变量,以微生物类群的丰度作为预测变量。因此,他们在肿瘤组织中确定了 2212 个显著且稳定性选择的宿主 CpG 甲基化-细菌关联,在 TCGA-CRC 队列的 AN 组织中确定了 1200 个此类关联(图S1G)。
他们将微生物相关的 CpG 位点注释到基因组区域。以总探针为背景,观察到微生物相关 CpG 在启动子相关区域富集,包括“TSS200”、“TSS1500”、“第 1 个外显子”和“5′ UTR”区域,但在 AN 组织中基因体和 3′ UTR 区域则耗尽。令人惊讶的是,这种富集在肿瘤组织中并不存在(图 5 B),表明肿瘤微环境中微生物启动子甲基化调控受到干扰。
接下来,他们重点研究了微生物与启动子 CpG 基因座之间的关联,发现队列中的 AN 和肿瘤组织中微生物分类群与 CpG 基因座之间分别存在 27 种和 41 种关联(图 5C-D)。值得注意的是,在 AN 组织中,观察到了有益健康的细菌与基因甲基化之间的多种关联(图 5C)。例如, Faecalibacterium prausnitzii与PPFIA2、NR3C1和EXO1的甲基化相关。特别是,EXO1 启动子的甲基化与F. prausnitzii和Faecalibacterium属的丰度呈正相关。
与 NIPSNAP2 启动子内的多个甲基化位点(chr7: 55963861 、chr7:55963871 和 chr7:55963874)表现出强烈的负相关性。NIPSNAP2 是一种线粒体膜蛋白,作为线粒体自噬受体,线粒体自噬缺陷与包括 CRC 在内的各种疾病的相关性越来越强。这些例子表明某些有益细菌可能通过表观遗传调控正常组织中的基因表达在维持正常细胞功能方面发挥作用。
相反,在肿瘤组织中,观察到更多与潜在机会性致病菌的关联(图 5D)。例如,脆弱拟杆菌(B . fragilis)与 C1D 甲基化呈正相关,而新生梭菌与 UNC5D 启动子甲基化呈正相关。令人印象深刻的是,最近在 CRC 中发现的致病菌Clostridium hathewayi与 SLC24A5 和 ADHFE1 的启动子甲基化呈关联。
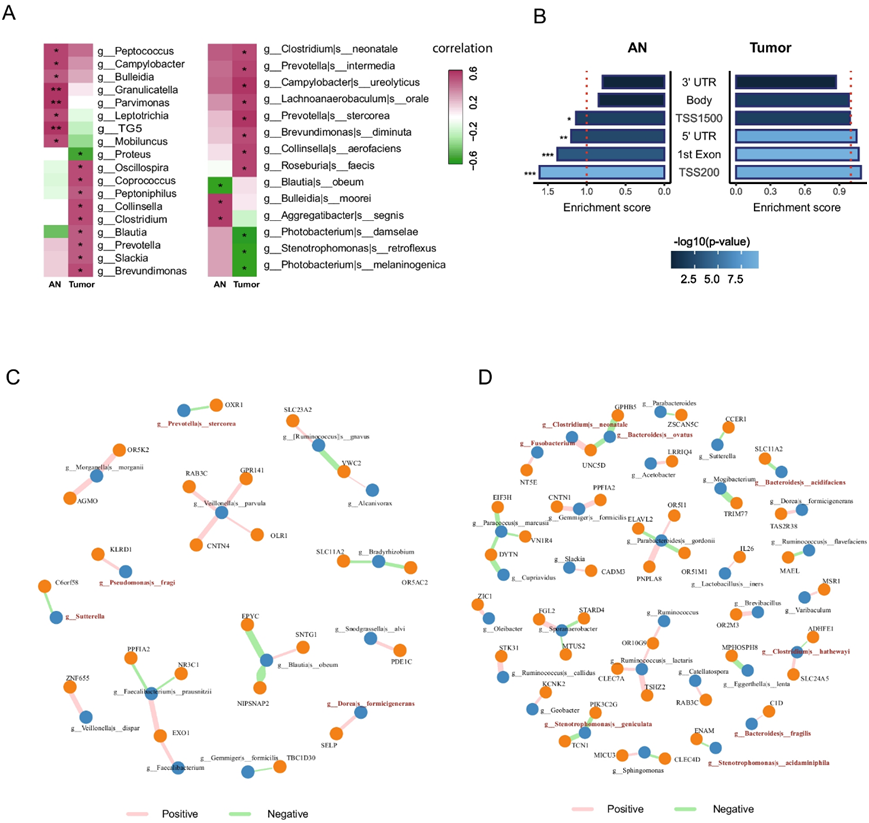
图5. 组织内细菌与宿主 DNA 甲基化的相互作用。
(A) 属(左图)和种(右图)水平的细菌丰度与 AN 和肿瘤组织中的总 DNA 甲基化水平之间的相关性。(B) 不同基因组区域中细菌相关甲基化位点的富集。(c-d) 临床样本的AN和肿瘤组织中细菌丰度与启动子 CpG 位点之间的关联。
06
肿瘤细菌经常与宿主基因甲基化相关
他们继续研究了 TCGA-CRC 队列和本研究数据中微生物与启动子 CpG 甲基化水平之间的关联,发现有五个属与肿瘤组织中的宿主启动子 CpG 甲基化水平表现出一致的关联。这些属包括梭杆菌属、寡养单胞菌属、拟杆菌属、青枯菌属和梭菌(图 6A)。相反,在两个数据集中,Sutterrella、多尔菌属、假单胞菌属、棒状杆菌属和普雷沃菌属的丰度与 AN 组织中的 CpG 甲基化水平相关(图 6B)。结合 TCGA-CRC 和本研究数据集中与这些分类群相关的基因甲基化,观察到肿瘤组织中微生物相关基因中 MAPK 级联、离子转运、凋亡过程和神经活性配体-受体相互作用相关功能的富集(图 6C)。相反,在 AN 组织中,基因主要富集碳水化合物结合功能(图 6D)。
梭杆菌和梭菌是与 CRC 进展相关的已知分类群。梭杆菌的丰度与 TCGA-CRC 队列中 C5AR1 启动子 (cg10224107) 的甲基化水平以及临床样本中 NT5E 启动子 (chr6:85449354) 的甲基化水平呈正相关。C5aR1 通过免疫调节在结直肠肿瘤发生中起主要调节作用。C5a/C5aR1 信号转导将髓系抑制细胞 (MDSC) 募集到发炎的结直肠,损害 CD8+ T 细胞并调节关键细胞因子和趋化因子的产生,从而引发 CRC。NT5E (CD73) 是一种抑制性免疫检查点分子,可抑制细胞免疫反应。梭杆菌丰度与 C5AR1 启动子高甲基化水平之间的正相关关系与具核梭杆菌在调节肿瘤免疫反应中的潜在作用相一致,例如创造有利于结直肠肿瘤进展的促炎微环境和增强检查点抑制剂阻断疗法的疗效。
他们进一步研究了微生物通路与宿主基因启动子甲基化状态之间的复杂联系。与肿瘤组织相比,AN组织中的相关性更加复杂。例如,“UDP-N-乙酰葡萄糖胺衍生的 O 抗原构建块生物合成通路”与多个基因的启动子甲基化模式相关,包括 C12orf45、GJD4、PNPLA8、MIR548X2 和 LINC00380。
他们建立了粪便和组织微生物组中细菌功能之间的相互关系(图7A-B)。随后,扩展了分析,以探索粪便微生物功能的丰度与肿瘤组织内 DNA 甲基化之间的关联。结果揭示了通路丰度与基因启动子甲基化水平之间存在负关联的三个实例。这些实例包括 L-谷氨酸降解 VIII 通路和 OR8G5、L-鼠李糖降解 I 通路和 C1D,以及嘧啶核碱基挽救和 NPFFR2通路。总的来说,这些发现强调了微生物组可能通过多种方式影响宿主 DNA 甲基化。
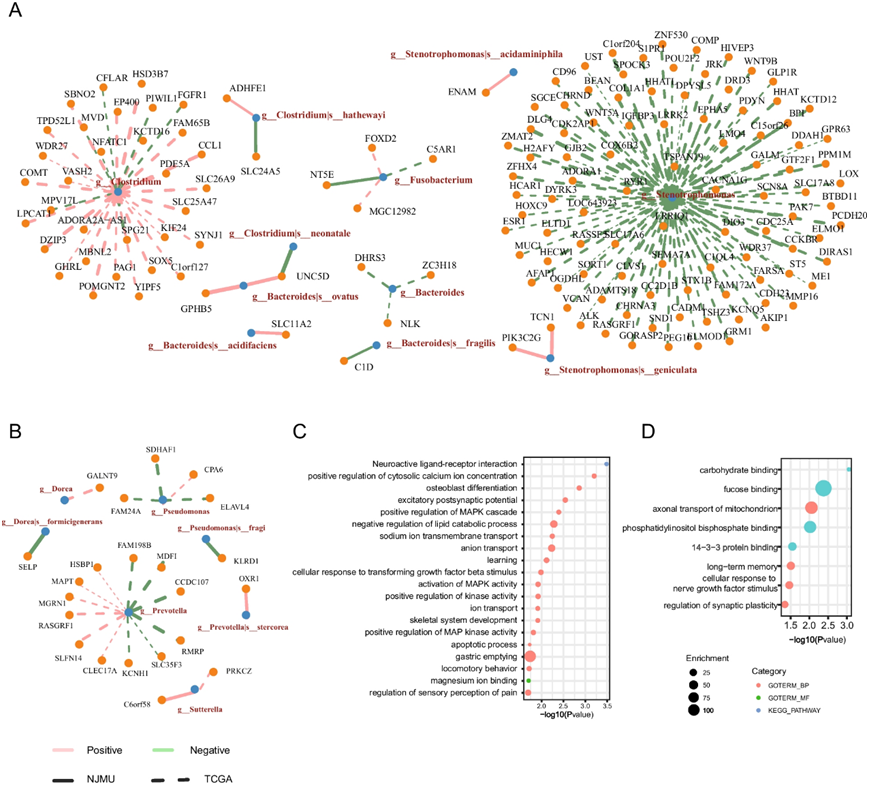
图6. 与宿主基因甲基化反复相关的分类群。
(a-b) TCGA-CRC 队列和本项研究数据(NJMU)中肿瘤和 AN组织中与宿主启动子 CpG 甲基化水平通常相关的分类群。(c-d) AN和肿瘤组织中微生物相关基因的功能富集。
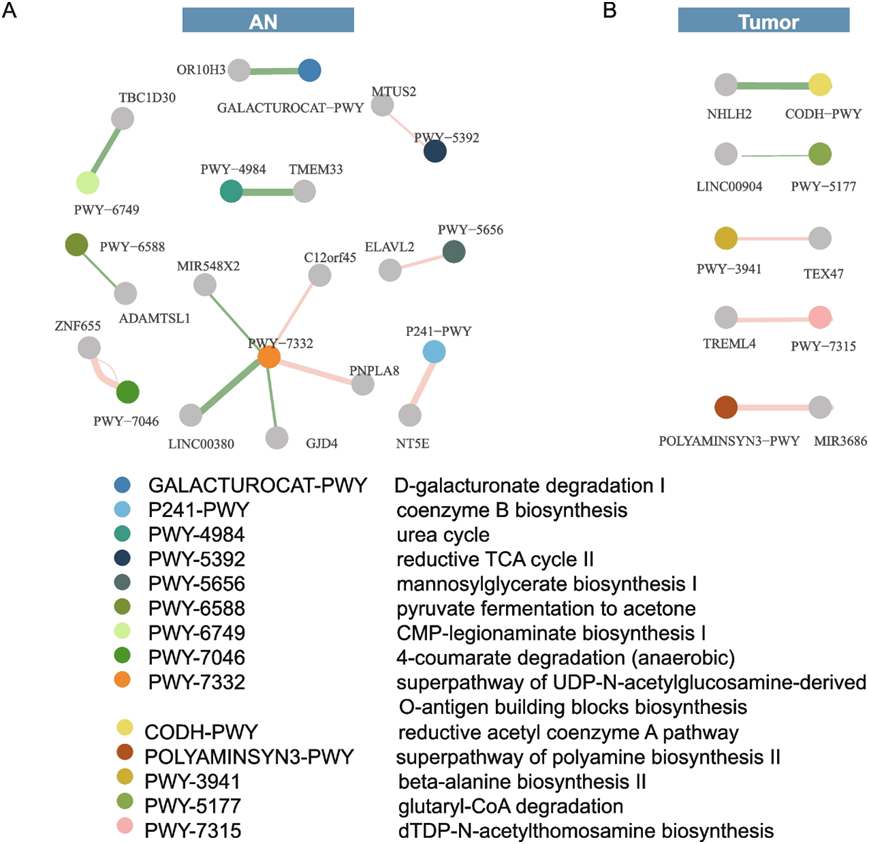
图7. 微生物与微生物通路和宿主DNA甲基化的相互作用。
(a-b) AN和肿瘤组织中与宿主基因甲基化水平相关的微生物通路。
+ + + + + + + + + + +
结 论
本项研究的分析显示,与邻近正常组织相比,CRC 组织的 DNA 甲基化组发生了显著改变。一项广泛的Meta分析结合了公开数据和内部数据,确定了肿瘤和邻近正常组织之间微生物衍生的甲基供体相关通路的显著变化。值得注意的是,观察到邻近正常组织中基因启动子区域内微生物相关 CpG 明显富集,而这种现象在肿瘤组织中明显不存在。此外,本项研究建立了肿瘤相关基因甲基化模式与特定细菌类群之间的一致且反复出现的关联。这项研究强调了肠道微生物群和致病菌在动态塑造 DNA 甲基化模式、影响生理稳态和促成 CRC 肿瘤形成方面的关键作用。这些结果为了解 CRC 发展中复杂的宿主-环境相互作用提供了宝贵的见解,并为这种疾病的治疗干预提供了新的理论依据。
+ + + + +



