

 English
English文献解读|Cell Rep Med(14.3):微生物和代谢谱揭示了肥胖相关结直肠癌中微生物与微生物之间的相互作用
✦ +
+
论文ID
原名:Microbial and metabolic profiles unveil mutualistic microbe-microbe interaction in obesity-related colorectal cancer
译名:微生物和代谢谱揭示了肥胖相关结直肠癌中微生物与微生物之间的相互作用
期刊:Cell Reports Medicine
影响因子:14.3
发表时间:2024.02.19
DOI号:10.1016/j.xcrm.2024.101429
背 景
肥胖是结直肠癌 (CRC) 的危险因素,肠道菌群参与肥胖和结直肠癌的发病机制已得到广泛认可。然而,区分肥胖相关结直肠癌和肥胖患者的粪便微生物组和代谢组的情况仍然未知。
实验设计
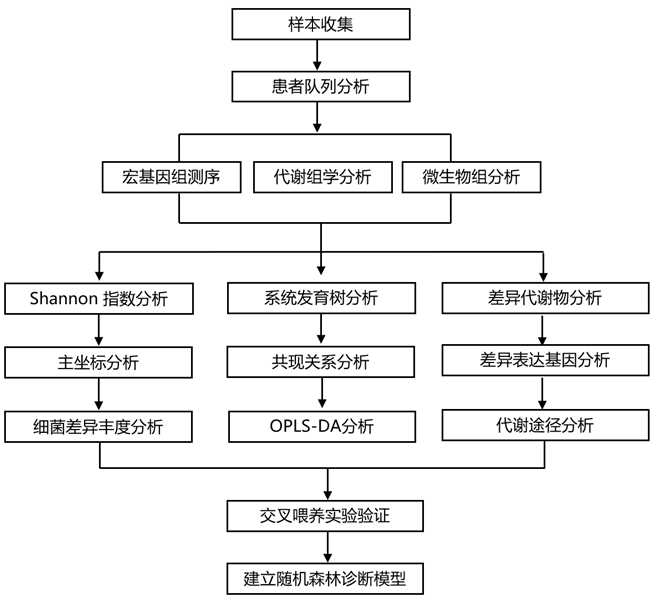
结 果
01

发现队列和验证队列概述
2018年至2021年从复旦大学附属肿瘤医院(中国上海)和山东大学第二医院(中国山东)收集了539份粪便样本。根据BMI标准将入组的CRC患者和健康对照分为四组:体重不足组(BMI≤18.5)、正常组(18.5<bmi≤24< span="">)、超重组(24<bmi≤28< span="">)、肥胖组(BMI>28)。
在发现队列中,418 名受试者由 CRC 患者和具有不同 BMI 组的健康对照组成,包括正常 CRC (N-CR)、超重 CRC (Ov-CRC)、肥胖 CRC (Ob-CRC)、正常健康对照(N-CTRL)、超重健康对照(Ov-CTRL)和肥胖健康对照(Ob-CTRL)。对发现队列的粪便样本进行宏基因组测序和代谢组学分析,以表征不同BMI组的肠道微生物分类和代谢特征,并研究差异物种、代谢物和微生物直系同源(KO)基因之间的关联(图1A)。在正常体重、超重和肥胖组中,与 CTRL 患者相比,在 CRC 患者中观察到通过 Shannon 指数测量的 α 多样性下降。与 N-CTRL 相比,N-CRC 的 α 多样性显著降低(图1B)。通过Bray-Curtis距离计算的主坐标分析(PCoA)显示群落组成的变化,不同BMI组的CRC和CTRL之间存在显著差异(图1C)。还对粪便代谢物进行了 PCoA,并显示三个 BMI 组之间的一致变化(图1D)。总的来说,这些数据表明不同 BMI 组的 CRC 与相应的 CTRL 具有不同的多样性和微生物距离指标,并且 Ov-CRC 和 Ob-CRC 显示出 α 多样性变化减少。
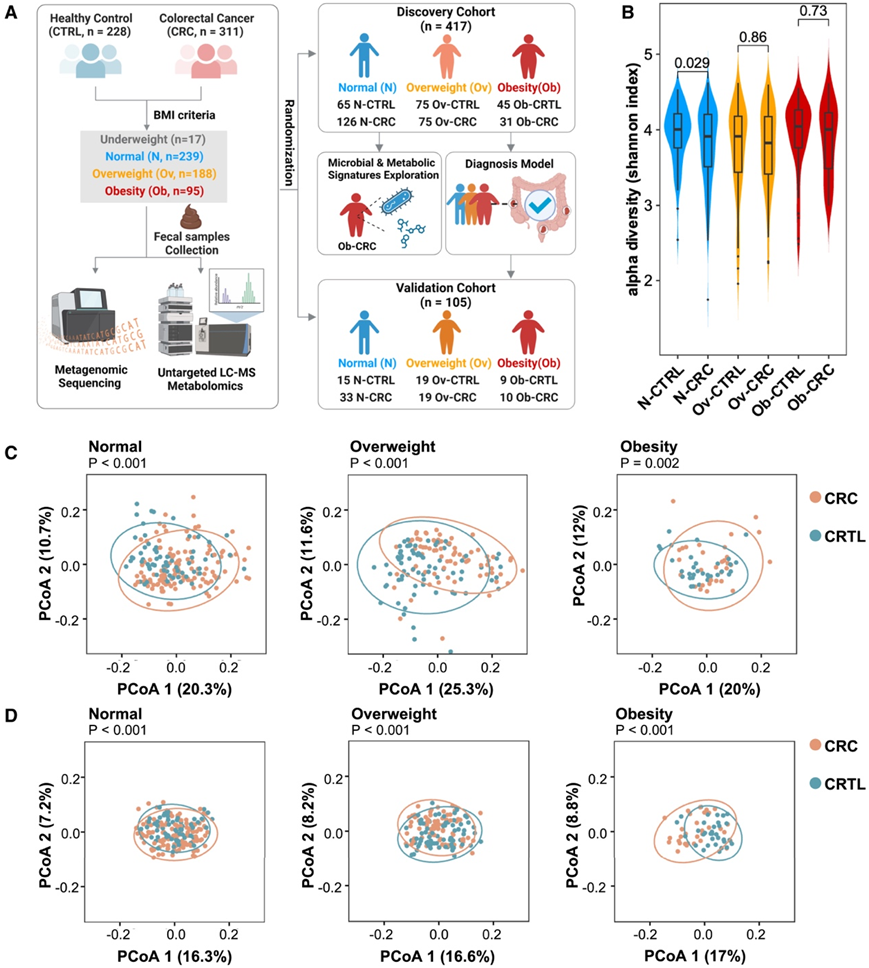
图1. 研究设计以及粪便样本的整体宏基因组和代谢组特征。
(A)共收集539份粪便样本,其中包括228名健康对照(CTRL)和311名结直肠癌(CRC)患者,根据BMI分为四组:体重不足组(灰色)、正常组(蓝色)、超重组(橙色)和肥胖组(红色)。(B) 通过Shannon指数检测不同组中 CRC 患者和健康对照的α 多样性。(C-D) 物种和代谢物的主坐标分析 (PCoA)。
02
Ob-CRC 和非肥胖 CRC 的微生物分类特征
他们鉴定了不同BMI组中CRC和CTRL之间差异富集的物种,并在N-CRC和Ov-CRC中发现了更多差异物种,它们与Ob-CRC丰度不同(图S1 A-C),显示了由 296 种细菌组成的差异丰度特征(图 2A)。
值得注意的是,与相应的CTRL相比,CRC中Peptostreptococcus stomatis和F. nucleatum的丰度在三组间均呈增加趋势。相关分析显示,在CTRL组中,P. stomatis的丰度与BMI呈负相关,而在CRC组中则呈正相关(图S1D)。F. nucleatum是一种机会性致瘤病原体。然而,在CRC或CTRL中未观察到F. nucleatum与BMI的相关性(图S1D)。在所有BMI组中,一些益生菌物种在结直肠癌中表现出一致的减少,例如Faecalibacterium prausnitzii和Dorea longicatena。相比之下,某些物种仅在N-CRC和Ov-CRC中表现出改变,而在Ob-CRC中保持不变,特别是包括Roseburia inulinivorans、Ruminococcus lactaris和Ruminococcus lactaris。F. prausnitzii作为一种益生菌,可以通过降解纤维和产生丁酸盐来调节能量摄入并发挥抗炎作用。F. prausnitzii在CRC和CTRL中都与BMI呈正相关(图S1D)。
此外,他们对鉴定的微生物标记物进行了共现分析(co-occurrence analysis),并探索了细菌间潜在的相互作用(图2B)。在3个BMI组中,F. prausnitzii、R. inulinivorans和D. formicigenerans之间存在稳定的共生关系。有趣的是,肿瘤富集物种(包括P. stomatis)和肿瘤耗尽物种(F. prausnitzii)之间的竞争关系仅在正常体重组和超重组中观察到。这些结果表明,不同BMI组CRC的独特微生物特征和细菌相互作用可能与Ob-CRC的肿瘤发展机制有关。
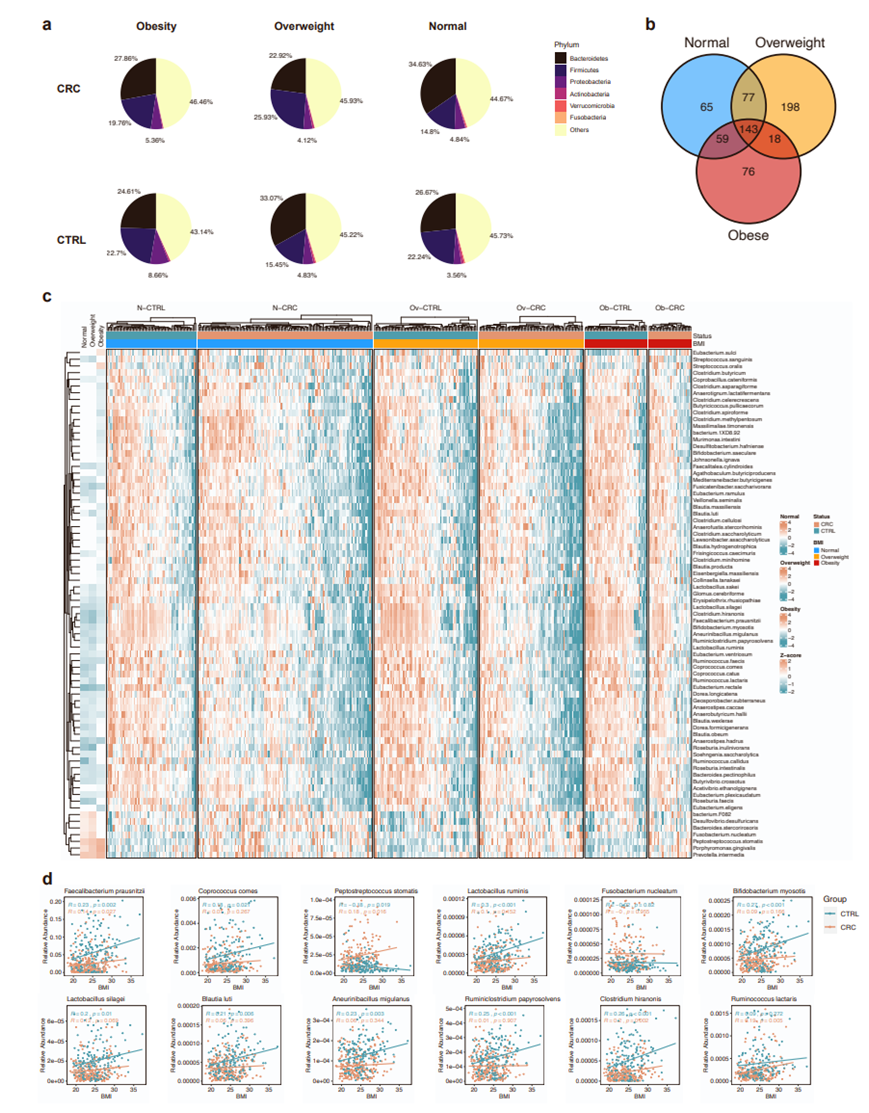
图S1. 不同BMI组CRC和CTRL的整体宏基因组特征。
(A)饼状图显示了不同BMI组CRC和CTRL在门水平上的微生物组成。(B)维恩图显示正常体重组、超重组和肥胖组差异种数。(C)已鉴定的差异细菌物种相对丰度热图。(D) CRC和CTRL区12种差异细菌物种相对丰度和BMI的散点图和线性拟合。
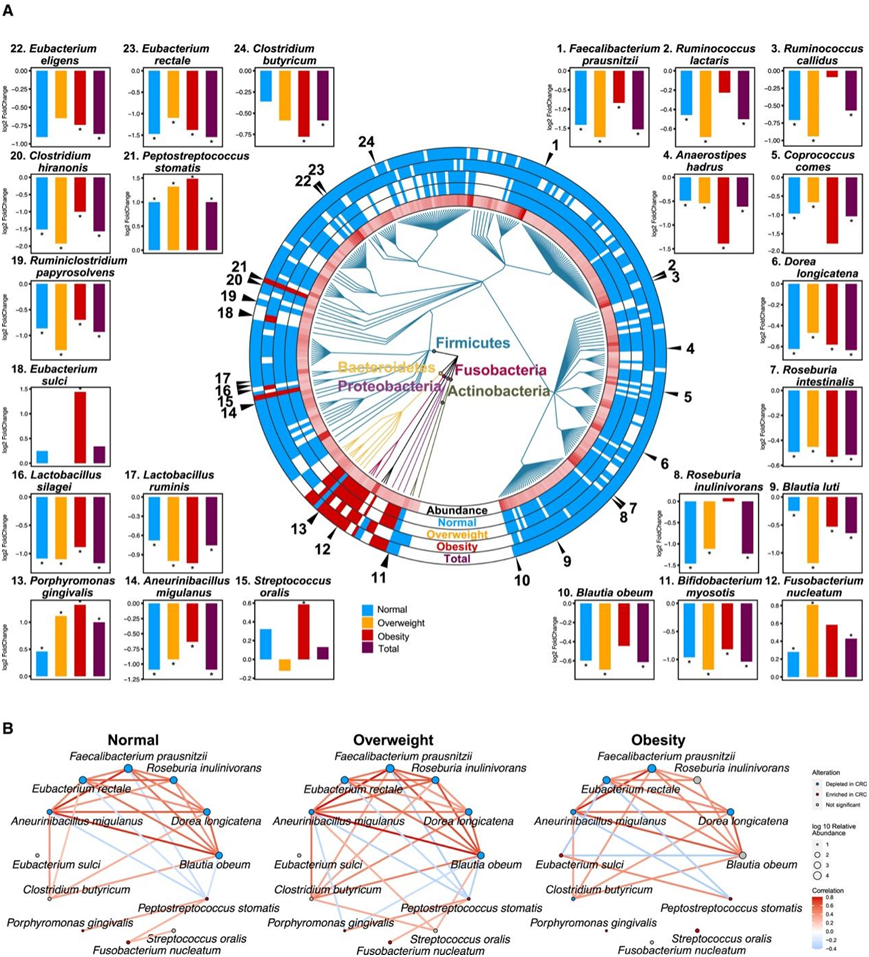
图2. 不同BMI组的差异物种以及CRC患者或健康对照的特定物种与BMI之间的关系。
(A) 与健康对照相比,三个 BMI 组的 296 种差异富集的细菌物种显示在系统发育树中,分为厚壁菌门、拟杆菌门、变形菌门、放线菌门和梭杆菌门。(B) 通过 Spearman 相关性得出正常体重、超重和肥胖组物种之间的潜在联系。
03
不同BMI组结直肠癌的粪便代谢组学改变
鉴于肠道微生物群与宿主-微生物共代谢之间的相互作用,他们对粪便样本进行了非靶向代谢组学研究,以确定不同BMI组CRC患者和健康对照组之间粪便代谢物的差异。正交偏最小二乘判别分析模型显示,各组CRC的代谢组成主要区别于相应的对照组(图3A)。然后,他们分别研究了N-CRC、Ov-CRC和Ob-CRC的差异代谢物(图3B)。大多数差异代谢物在肥胖组和非肥胖组之间是不同的。在这项分析中,与Ob-CTRL相比,Ob-CRC的代谢物变化特别大,包括富集的琥珀酸和丙酸以及缺失的十二烷二酸。与脂肪酸代谢密切相关,在Ob-CRC中观察到参与磷脂代谢的代谢物,包括丰富的溶血磷脂酰甘油(LysoPG) 14:1和耗竭的plasmenyl-磷脂酰胆碱(PC)(plasmenyl-PC) 29:0。磷脂降解产生的LysoPG和游离脂肪酸增加会产生更多的花生四烯酸和类二十烷酸,从而导致炎症和促进肿瘤的微环境。Plasmenyl-PC是一种与肥胖和调节氧化应激负相关的缩醛磷脂。与Ob-CTRL相比,Ob-CRC中plasmenyl-PC进一步减少,表明氧化应激升高是肥胖人群CRC发生的关键机制。在CRC和CTRL中,胆固醇与BMI呈负相关,与相应的健康对照相比,仅在N-CRC和Ov-CRC中发生改变。
他们进一步研究了在不同BMI组的CRC患者和健康对照中所选择的差异物种与差异代谢物之间的相关性(图3C)。例如,F. prausnitzii与Ob-CTRL中的胆固醇呈负相关,提示F. prausnitzii可能参与胆汁酸代谢。他们还验证了F. prausnitzii体外代谢胆固醇的能力。这些有机化合物分别在Ob-CRC和非肥胖型CRC中的独特作用仍需进一步研究,可能参与CRC中独特的代谢-微生物相互作用。
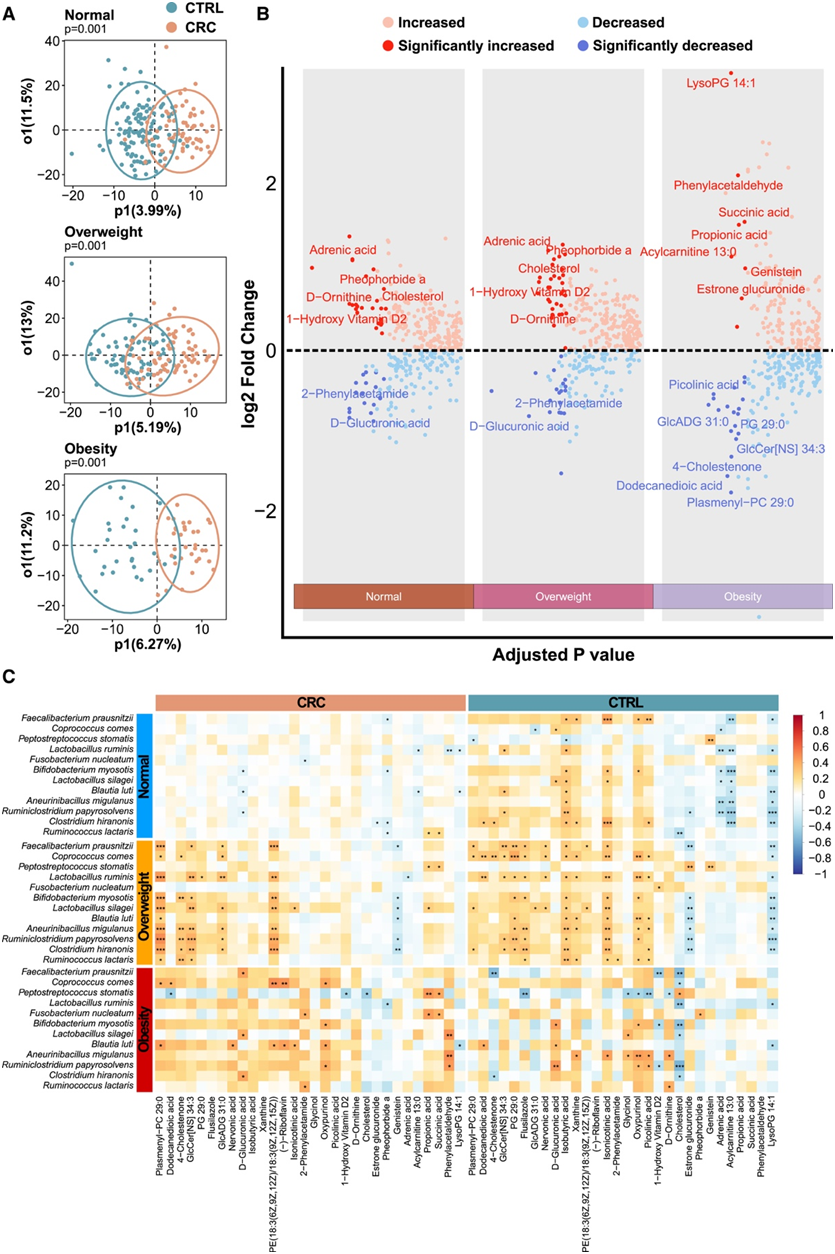
图3. 不同BMI组CRC患者和健康对照的代谢组学特征。
(A) 每组 CRC 患者和健康对照代谢物的正交偏最小二乘判别分析 (OPLS-DA)。(B) 每组差异代谢物的火山图。(C) 正常体重、超重和肥胖组的 CRC 和 CTRL 中差异物种和代谢物之间的 Spearman 相关性。
04
不同BMI组CRC中KO基因和KEGG通路中微生物基因的改变
鉴于结直肠癌肠道微生物组和代谢组的多组学转变,他们假设代谢物差异可能反映了不同BMI组结直肠癌患者微生物酶基因表达的差异。为了进一步确定 Ob-CRC 和非肥胖 CRC 中发生的微生物代谢过程,他们对 KO 数据库中的微生物基因进行了注释,并绘制了热图以显示相关途径中分组的差异表达 KO 基因(图4A)。Ob-CRC 中涉及甲烷代谢、甘油磷脂代谢和脂多糖 (LPS) 生物合成的途径发生显著改变。与甲烷代谢相关的富集 KO 基因(torA、torC、torD、torY和torZ)编码三甲胺 N-氧化物 (TMAO) 还原酶,负责将 TMAO 转化为三甲胺 (TMA),这在电子传递中起着至关重要的作用链。这一结果表明 Ob-CRC 背景下 TMAO 代谢可能受到破坏。KO 基因pldA编码一种磷脂酶,该酶在甘油磷脂代谢的各个方面发挥着关键作用,包括 PC、磷脂酰乙醇胺和磷脂酰丝氨酸的分解代谢。此外,glpB和glpC构成3-磷酸甘油脱氢酶的两个重要亚基。这些代谢物在 Ob-CRC 的背景下特别值得注意,因为它们可能会导致脂质稳态的破坏,这一观点通过观察到的粪便代谢物(包括 PG、LysoPG 和 plasmenyl-PC)的代谢扰动得到证实(图 3 B)。源自肠道微生物群的脂多糖是与炎症相关的内毒素,可增加肠道通透性。
为了验证观察到的 KO 基因丰度变化是否归因于特定细菌物种,他们对 KO 基因与这些物种之间的相关性进行了整合分析(图 4 B)。P. stomatis与torZ正相关,提示其可能参与TMAO代谢。益生菌如F. prausnitzii和R. inulinivorans与相当比例的KO基因缺失正相关,表明CRC中益生菌的缺乏可能是导致代谢失衡的关键因素。总的来说,这些研究结果表明Ob-CRC表现出独特的微生物酶促反应和代谢过程,由微生物基因谱的显著改变驱动。
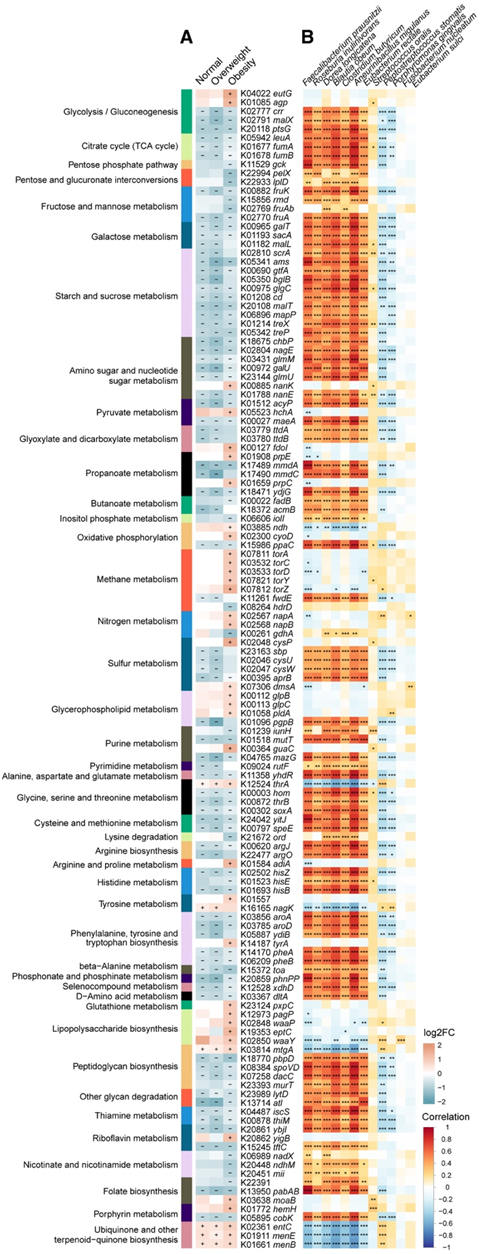
图4. KO基因中CRC相关的功能改变。
(A) 热图显示了综合Meta分析,该分析确定了在三个 BMI 组中的每个 KEGG 通路中 KO 基因表达的显著变化。(B) 差异物种和 KO 基因之间的 Spearman 相关性。
05
代表性标记物之间的微生物和代谢关联
为了剖析宿主和微生物群之间可能构成肥胖特征的相互作用,他们对所有差异富集的类群、代谢物和 KO 基因之间进行了 Spearman 相关性分析,并绘制了正常组、超重组和肥胖组中具有代表性特征的网络图(图 5A)。与其他两组相比,在肥胖组观察到复杂的网络。在肿瘤富集的P. stomatis与异丁酸和十二烷二酸等多种代谢物呈正相关,而F. prausnitzii与这些代谢物正相关。
接下来,他们在prausnitzii和P. stomatis之间进行了交叉饲养实验(图5B)。孵育48 h后,他们观察到在交叉饲养条件下F. prausnitzii和P. stomatis的生长均显著高于对照(图5C)。靶向代谢组学鉴定出了参与两种物种交叉取食关系的候选代谢物,包括乙酸、异丁酸、尿嘧啶和2-羟基己酸。基于这些结果,他们推测促癌细菌可能交叉喂养益生菌以获得生态位,从而在肥胖的背景下促进癌症的发展。
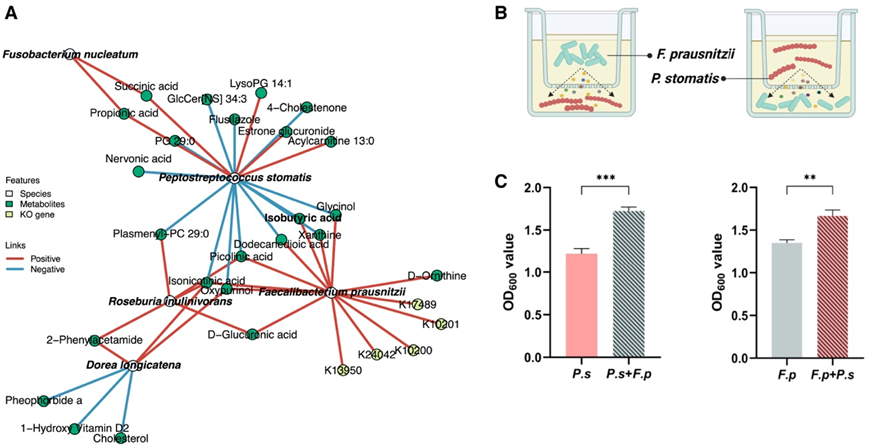
图5. 物种、KO基因和代谢物之间的整合网络以及肥胖组的交叉喂养图。
(A) 物种、KO 基因和代谢物之间的整合网络。(B) 示意图说明了F. prausnitzii和P. stomatis之间交叉喂养实验的设计。(C)与单一培养对照相比,交叉饲喂条件下F. prausnitzii和P. stomatis的细菌浓度。
06
不同BMI组结直肠癌随机森林诊断模型
不同BMI组的CRC患者的差异物种和代谢物存在差异,因此他们假设CRC风险较高的肥胖人群应该使用独立的诊断模型来更准确地筛查CRC。为了研究微生物和代谢谱作为诊断标记的潜力,他们建立了随机森林分类器,以区分同一BMI组和不同BMI组的健康对照中N-CRC、Ov-CRC和Ob-CRC病例(图6)。在发现队列中进行Boruta特征选择,选择关键的鉴别细菌分类群。三个BMI组选择的分类群不同,这验证了他们的假设,即不同BMI的结直肠癌患者需要不同的生物标志物和诊断模型(图6A)。
此外,将一个独立的外部验证队列分为正常组、超重组和肥胖组,用于验证每个分类器的诊断潜力。已成功构建的诊断分类器在正常、超重和肥胖组中的受试者工作特征曲线下面积 (AUROC) 分别为0.9354、0.8892和0.8(图6B)。在超重和肥胖组中也进行了组间验证(图6C-D)。为了检验诊断策略的稳健性和有效性,他们进行了20次重复的组对组验证分析,并计算了平均AUROC(图6E)。
总体而言,从每组中选择的特征都显示出在同一BMI组中识别结直肠癌患者的出色能力,而在其他两个BMI组中识别结直肠癌患者的能力较差,表明其在不同BMI组人群的筛查策略中具有很大的潜力。
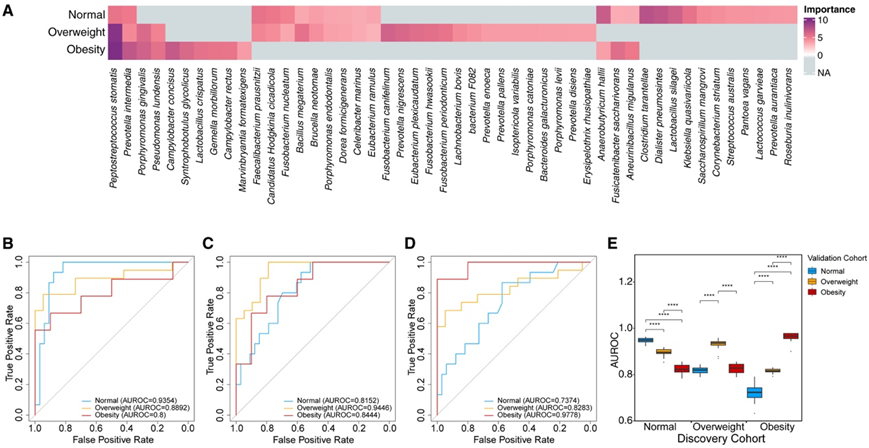
图6. 从三个BMI组中分别选择的微生物标志物提高了诊断效率。
(A) 发现队列中每个 BMI 组中选定微生物特征重要性的热图。(B) 由发现队列的 N-CRC 和 N-CTRL 构建的模型在验证队列的正常组、超重组和肥胖组中进行验证,并计算 AUROC 值。(C) 该模型是根据发现队列的 Ov-CRC 和 Ov-CTRL 构建的,并在验证队列的三组中的每一组中进行验证。(D) 该模型是根据发现队列的 Ob-CRC 和 Ob-CTRL 构建的,并在验证队列的三组中的每一组中进行验证。(E) 箱线图显示了使用 (A) 中选定特征的模型的组间验证的 AUROC 值。
+ + + + + + + + + + +
结 论
本项研究利用 522 名 CRC 患者和健康对照的宏基因组测序和代谢组学来确定肥胖 CRC 的特征。综合分析表明,与肥胖相关的结直肠癌的特点是Peptostreptococcus stomatis含量升高、脂肪酸和磷脂失调,以及涉及甘油磷脂代谢和脂多糖合成的途径改变。相关分析揭示了肥胖症中的微生物相互作用,其中益生菌Faecalibacterium prausnitzii和P. stomatis会促进肿瘤发生。体外实验验证了它们在交叉喂养条件下生长增强。微生物与微生物之间的相互作用可能有助于肥胖与结直肠癌风险升高之间的关联。此外,结合了 BMI 特异性微生物生物标志物的诊断模型有望实现精确的 CRC 筛查。
+ + + + +



