

 English
English文献解读|Nat Commun(16.6):纵向血浆蛋白质组分析揭示了结直肠癌诊断和西妥昔单抗治疗反应的生物标志物的多样性
✦ +
+
论文ID
原名:Longitudinal plasma proteome profiling reveals the diversity of biomarkers for diagnosis and cetuximab therapy response of colorectal cancer
译名:纵向血浆蛋白质组分析揭示了结直肠癌诊断和西妥昔单抗治疗反应的生物标志物的多样性
期刊:Nature Communications
影响因子:16.6
发表时间:2024.02.01
DOI号:10.1038/s41467-024-44911-1
背 景
西妥昔单抗(Cetuximab)疗法是结直肠癌(CRC)的主要治疗方法,但耐药性限制了其有效性。因此,有必要找到西妥昔单抗首次治疗反应的潜在预测生物标志物。
实验设计
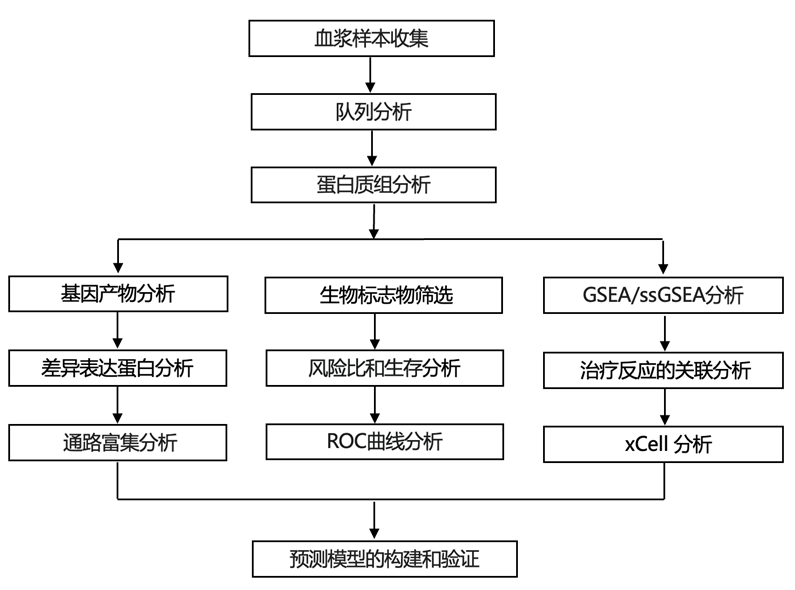
结 果
01

CRC 抗 EGFR 治疗队列血浆和组织蛋白质组分析的特征
为了研究 CRC 的蛋白质组模式以及与西妥昔单抗治疗反应的关联,研究团队从血浆队列中收集了 641 份血浆样本,该血浆队列由两个独立队列组成,包括发现队列和验证队列(图1A)。在血浆发现队列的所有血浆样本中共鉴定出 9852 个基因产物(GP),其中在 CRC 患者中鉴定出 9714 个 GP,在健康对照(HC)中鉴定出7512 个(图1B)。为了探索 CRC 患者与健康对照的分子差异,他们对治疗前 CRC和 HC 进行了比较蛋白质组分析。治疗前CRC和HC之间的蛋白质组覆盖率没有显著差异(图1C)。78.9% 的蛋白共同存在于治疗前的 CRC 和 HC 中,根据人类蛋白质图谱(HPA)数据库中的蛋白质类别将这些蛋白归类为分泌蛋白。重要的是,有13.2%的蛋白质(1138个糖蛋白)仅存在于治疗前的结直肠癌和健康对照中;其中,CRC相关蛋白所占比例较高,代表血浆蛋白质组最能反映CRC的分子改变(图 1C-D)。
为了检测治疗前 CRC 患者和健康对照之间血浆蛋白质组的变化,他们比较了 CRC 和 HC 组的蛋白质组图谱,共鉴定了 1269 个差异表达蛋白(DEP),其中 745 个蛋白质在治疗前上调,524 个蛋白在HC 组中上调(图1E)。
然后,他们根据从31个不同的公共数据库获得的ConsensusPathDB (CPDB)分子相互作用数据,基于DEP进行途径富集分析。他们发现糖酵解/糖异生、ERBB2信号传导、细胞对化学应激的反应、MAPK激活和细胞外基质组织途径在结直肠癌患者的血浆样本中富集(图1F-G)。健康人血浆标本均有中性粒细胞脱粒、先天免疫系统等特征(图1F-G)。
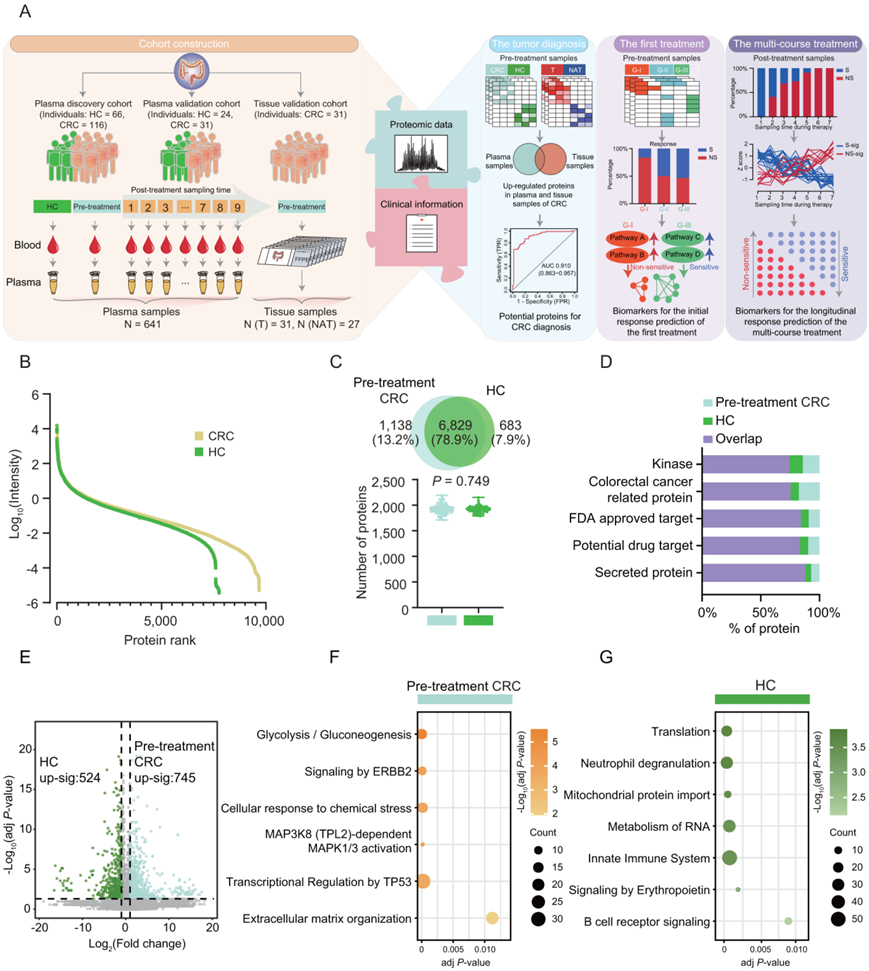
图1. CRC 队列和健康对照的血浆蛋白质组分析摘要。
(A) 血浆蛋白质组实验流程概述。(B) 根据 CRC 和 HC 中蛋白质丰度降序排列的 CRC 队列和健康对照的蛋白质鉴定的动态范围。(C) 维恩图显示治疗前 CRC 和 HC 的蛋白质重叠。 (D) 条形图显示治疗前 CRC 患者和健康对照 (HC) 中的蛋白质比例。 (E) 火山图显示治疗前 CRC 队列和健康对照的差异表达。(F-G) 气泡图显示治疗前 CRC和 HC组的 CPDB 通路富集。
02
用于诊断 CRC 患者的血浆蛋白生物标志物
为了寻找可用于血浆和组织样本的潜在蛋白质,作为区分结直肠癌患者与健康对照的生物标志物,他们在血浆和组织样本中采用了严格的筛选策略,如下(图 2A):(1)候选蛋白质至少 50% 的样品中有蛋白质表达;(2)候选者在肿瘤样本中显著高于正常样本;(3) 候选者的 CRC 样本数比正常样本或正常邻近组织 (NAT) 至少增加 2 倍。分别筛选出148个和797个显著且稳定过表达的蛋白;其中,CRC患者血浆和组织样本中重叠的15个特征(CPT1A、NUP205、CDC37、MAT2A、RPN1、GMPS、PSMA1、CDH1、SRSF7、FUBP3、PIGR、S100A8、S100A9、THBS2和COL12A1)均升高。结合三个独立队列,他们最终确定了一组蛋白质:包含了COL12A1、THBS2、S100A8和S100A9,这些蛋白在CRC患者的血浆和组织中均显著增加(图2B)。
然后他们进行了基因集富集分析(GSEA),探索这4种蛋白(COL12A1、THBS2、S100A8和S100A9)的潜在生物学关联。GSEA结果显示,根据Gene Ontology注释,基于细胞成分数据库,在含胶原的细胞外基质(ECM)(GO:0062023)中,COL12A1、THBS2、S100A8和S100A9四个蛋白富集(图2C)。蛋白质-蛋白质相互作用网络揭示了参与 ECM 的这些蛋白质之间的密切联系,特别是 COL12A1、THBS2、S100A8 和 S100A9 四种蛋白质(图 2C)。根据HPA注释,这4种蛋白属于分泌蛋白,其中,S100A8和S100A9作为潜在的标记物和抑制剂具有临床实用性(图 2D)。
为了进一步确定这四种蛋白质是否能够有效地区分 CRC 患者与健康对照,他们使用机器学习算法来评估这些蛋白质的预测能力,计算了血浆发现队列中四种蛋白质的受试者工作特征 (ROC) 曲线图的曲线下面积 (AUC),发现单个标记物的 AUC 至少为 0.674(范围:0.674–0.843);组合血浆标志物的AUC增加至0.910(95%CI:0.863-0.957),表现出更好的区分CRC患者和健康对照的性能(图 2E-F)。更重要的是,四种蛋白质的组合预测在独立组织验证队列中取得了良好的性能,AUC为0.945(图 2G)。为了进一步验证四种蛋白质区分 CRC 患者和健康对照的预测效果,他们纳入了由 31 名 CRC 患者和 24 名健康对照组成的独立血浆验证队列。结果表明,在独立血浆验证队列中,这四种蛋白可以很好地区分结直肠癌患者和健康对照,AUC为0.952(图 2G)。
他们根据血浆蛋白质组数据分别对20例CRC患者、RAS突变的CRC患者和20例RAS突变的CRC患者进行了差异分析和ROC分析。差异分析发现四种蛋白(COL12A1、THBS2、S100A8 和 S100A9)在总共 20 名 CRC 患者中表现出显著上调,ROC 分析表明这四种蛋白能够很好地区分 CRC 和 HC,AUC 为 0.948 (图 2H)。在RAS突变的CRC患者中,他们也发现这四种蛋白在RAS突变的CRC患者中表现出一致的上调;同时,四种蛋白质实现了较高的预测,AUC为0.985(图 2H)。
ROC分析表明,四种蛋白联合预测可以实现CRC诊断的高预测,AUC为0.917,表明四种蛋白在CRC诊断中的稳定性和普适性(图 2I)。在 CPTAC CRC 队列中,42%的 CRC 患者有RAS突变,90%的 CRC 患者没有肿瘤转移。为了进一步证明这四种蛋白的预测效率不仅限于RAS野生型转移性CRC患者,他们进一步对40名具有RAS突变的CRC患者进行了分层,其中ROC分析显示其与肿瘤和NAT有很好的区分,AUC为0.912(图 2I)。此外,他们还对86名非转移性CRC患者进行了分层,其中ROC分析也显示出良好的预测,AUC为0.931(图 2I)。因此,他们验证了四种蛋白对更广泛人群规模的CRC患者的预测作用,但不限于RAS野生型CRC患者或转移性CRC患者。
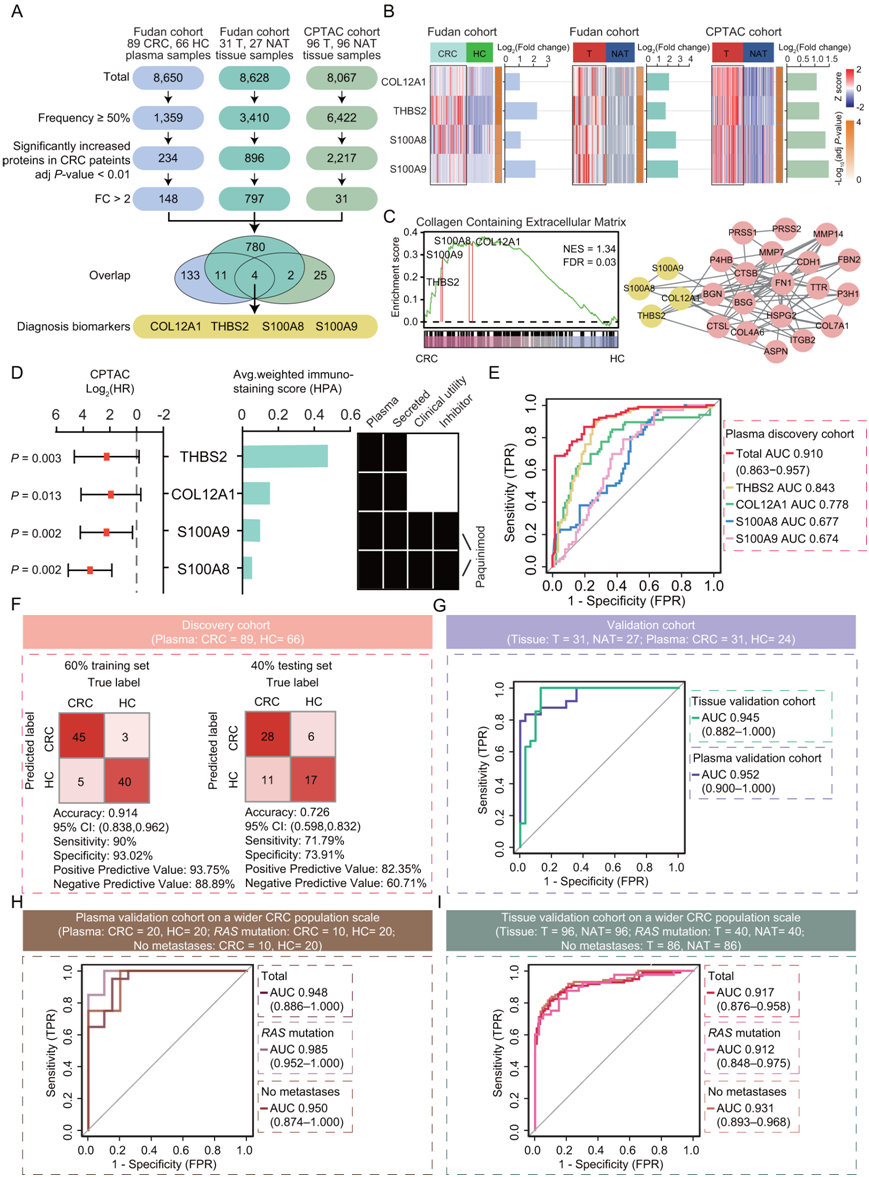
图2. 用于诊断 CRC 患者的血浆蛋白生物标志物。
(A) CRC诊断生物标志物筛选标准适用于复旦队列和CPTAC CRC队列。 (B) 热图显示了复旦队列和 CPTAC CRC 队列中四种蛋白质的相对丰度。(C) 左:GSEA 显示 CRC 患者体内富含含有细胞外基质的胶原蛋白。右:蛋白质-蛋白质相互作用网络。 (D) 左:CPTAC 队列中总体生存的风险比 (HR)和由这些诊断的人类蛋白质图谱 (HPA) 定义的免疫组织化学 (IHC) 染色评分生物标志物。 (E) 四种蛋白质的受试者工作特征 (ROC) 曲线,用于区分 CRC 和 HC。 (F) 使用 60% 训练集和 40% 测试集的逻辑回归分类器的分类误差矩阵,用于区分血浆发现队列中的 CRC 和 HC。 (G) 独立组织验证队列和血浆验证队列中四种蛋白质的 ROC 曲线。 (H-I) 在更大的CRC人群规模上,复旦血浆验证队列和CPTAC组织验证队列中区分CRC患者与HC或NAT的四种蛋白的ROC曲线。
03
西妥昔单抗治疗初始反应的潜在分子特征和生物标志物
为了探索预测治疗反应的潜在生物标志物,他们关注了 89 名初治 CRC 患者的基线血浆蛋白质组谱,并选择客观缓解率(ORR) 来进行疗效评价,ORR可分为部分缓解(PR)和完全缓解(CR);CR和PR的患者定义为敏感(S),疾病稳定(SD)和疾病进展(PD)的患者定义为不敏感(NS)。他们首先对89 个未接受治疗的血浆样本进行共识聚类分析,鉴定了三种亚型:G-I、G-II和G-III。对三种蛋白质组亚型中所有这些临床特征的进一步统计分析揭示了蛋白质组亚型与治疗反应的显著相关性(图3A),但在肿瘤的任何级别、程度中均未观察到这种相关性。这些结果表明,蛋白质组亚型可以反映与治疗反应的强烈相关性,而与其他临床参数无关。
治疗反应表现出从GI到G-III逐渐降低的耐药性现象,敏感患者(S:CR和PR)的比例从G-I的16.7%显著增加到G-III的53.6%,而非敏感患者(NS:SD和PD)的比例则从G-I的83.3%显著减少到G-III的46.4%(图 3A)。
基于单样本基因集富集分析 (ssGSEA)评分的进一步分析表明,在这些富含G-III亚型的通路中,自噬与中性粒细胞脱颗粒或先天免疫系统显著相关,表明自噬与中性粒细胞脱颗粒和先天免疫系统的生物学关联(图3B)。他们发现G-III亚型与其他两种亚型相比具有最高的免疫评分(图 3C),并且CD8+ Tem在G-III亚型中显著富集(图 3D)。
为了探讨这些细胞类型与治疗反应的关联,他们比较了 S 组和 NS 组之间的 xCell 评分,他们发现CD8+Tem的xCell评分在S组中显著占优势(图 3E)。为了验证这一发现,他们对由 12 名 S 患者和 19 名 NS 患者组成的独立组织队列的组织蛋白质组数据进行了 xCell 分析。一致地,基于组织蛋白质组,与NS组相比,S组中CD8+Tem的xCell评分显著升高(图 3E)。结果,S组CD44和GZMK的表达明显高于NS组。此外,S组CD44阳性细胞比例和GZMK阳性细胞比例高于NS组(图3F)。
为了进一步探讨CD8 Tem的临床意义,他们进一步将CD8+ Tem评分与CT/MRI评估的肿瘤大小联系起来。结果表明,CD8+ Tem xCell评分较高的患者在基线评估中往往具有较小的肿瘤大小(图3G)。进一步的相关分析显示,肿瘤大小与CD8+ Tem xCell评分之间存在显著的负相关(图3H),提示CD8 Tem可能是西妥昔单抗敏感性的潜在标志物。
然后,他们研究了与治疗反应相关的潜在生物相互作用。据报道,自噬已纳入多种先天性和适应性免疫途径。越来越多的证据表明,自噬在调节中性粒细胞功能(包括脱颗粒、代谢和 NET 形成)方面具有重要作用。自噬缺陷会破坏中性粒细胞脱颗粒。他们提出了潜在的综合网络图,其中参与自噬、中性粒细胞脱粒和T细胞活化的相关蛋白在S组中占主导地位(图 3I)。在这些蛋白中,血浆和组织样本中的RPTOR和IMPDH2与CD8+Tem均呈显著正相关(图 3J),并且RPTOR的高表达和IMPDH2与经CPTAC队列验证的更好预后显著相关(图 3K)。此外,通过数据独立采集(DIA)策略确定的关键敏感调节因子(RPTOR和IMPDH2)通过平行反应监测(PRM)分析对来自血浆发现队列和另一个独立血浆验证队列的血浆样本进行了进一步验证(图 3L)。总体而言,自噬的高度激活和 CD8+ Tem 的聚集可能会改善对西妥昔单抗治疗的反应。
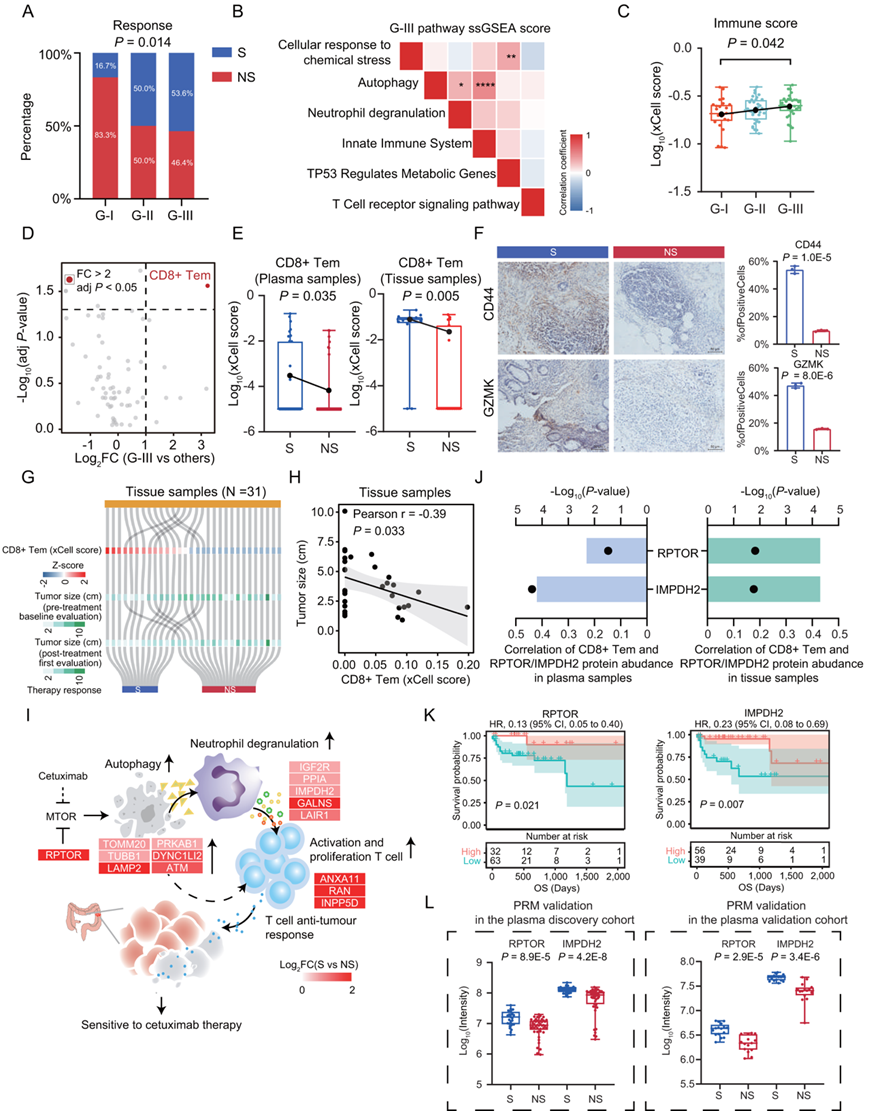
图3. 西妥昔单抗敏感性的潜在机制和生物标志物。
(A) 蛋白质组亚型与治疗反应的关联。 (B) G-III 亚型路径的相关性。 (C,E) G-I、G-II、G-III组血浆和组织样本免疫评分以及S组和NS组CD8+Tem评分的箱线图。(F) 代表性样品免疫组化染色CD44和GZMK的鉴定。 (G) CD8+Tem 评分(Z 评分)与肿瘤大小(cm)和 S/NS 组之间的关联。(H) CD8+Tem 评分与肿瘤大小的相关性。 (I) 显示西妥昔单抗敏感性的潜在机制的示意图。(J) CD8+Tem 评分与 RPTOR/IMPDH2 的相关性。(K)生存曲线分析。(L) RPTOR和IMPDH2在发现组和验证组中的差异表达。
04
西妥昔单抗耐药的潜在机制和生物标志物。
作为非敏感亚型,GI亚型NS患者比例最高(图 4A),其特点是PPAR-α通路、整合素相关激酶信号传导、维生素和辅因子代谢、ECM 、RHO GTPase 循环和 MAPK 信号通路(图 4B)。为了进一步确定富含 GI 亚型的通路与治疗反应的关联,他们在 S 组和 NS 组之间进行了 ssGSEA 分析。结果显示,NS 组 MAPK 信号、RHO GTPase 循环和 ECM 通路的 ssGSEA 通路评分显著升高,经 GSEA 验证,NS 组这些通路显著富集(图 4C-D),显示这些信号通路可能作为西妥昔单抗治疗无反应的指标。
在本研究中,他们发现RRAS和RRAS2高表达的CRC患者容易对西妥昔单抗治疗产生耐药(图 4E)。然后,他们想知道并探索伴随着RRAS/RRAS2的高表达,哪条下游通路发生激活。根据RRAS/RRAS2的平均表达量,将队列分为RRAS高表达组和RRAS低表达组。有趣的是,他们发现RRAS高表达组中MAPK信号、RHO和ECM通路的ssGSEA评分显著上调(图 4F),并通过GSEA验证(图4G)。他们提出了 RRAS/RRAS2 蛋白正向调节三个下游通路激活的潜在调控轴,这表明与西妥昔单抗治疗耐药相关(图4H)。
在参与这些通路的蛋白中,FBLN1、MMP8和ITGA5与RRAS/RRAS2呈显著正相关,并且FBLN1、MMP8和ITGA5的高表达与CPTAC队列中验证的较差预后显著相关(图4I-K)。这些结果表明,RRAS/RAS2 正向调节 ECM 通路,与西妥昔单抗治疗耐药以及 CRC 患者不良预后相关。ROC 分析显示,这些蛋白质的组合具有很高的准确性,AUC 为 0.849。为了进一步验证这种蛋白质组合的稳健性,他们在独立的组织和血浆验证队列中使用了机器学习算法。ROC 分析结果在区分 S 患者和 NS 患者方面表现良好,在组织和血浆验证队列中 AUC 值分别为 0.816和 0.890(图 4L)。
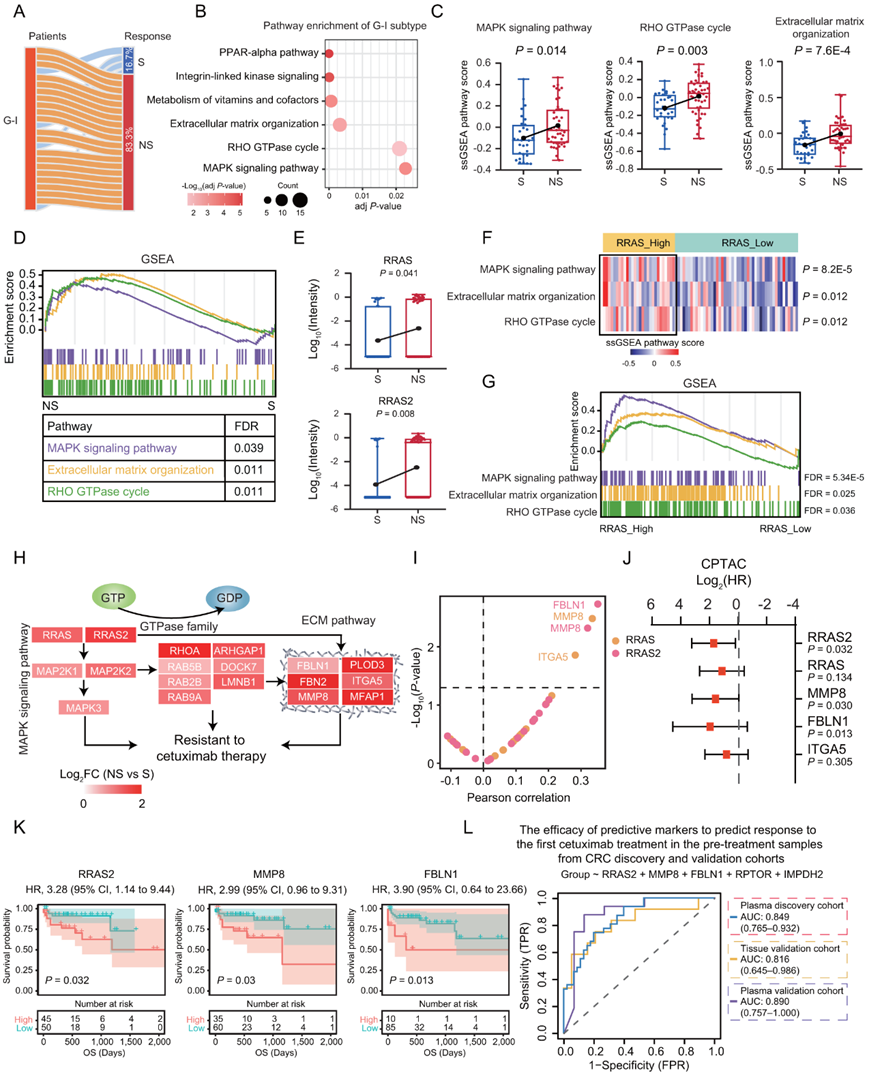
图4. 西妥昔单抗耐药的潜在机制和生物标志物。
(A) 胃肠道亚型中西妥昔单抗治疗的反应比例。(B) GI 亚型的 CPDB 通路富集。(C) S 组和 NS 组之间路径 ssGSEA 评分的箱线图。 (D) 与 S 组相比,NS 组中 MAPK 信号、RHO GTPase 循环和 ECM 通路的 GSEA 富集。 (E) S 组和 NS 组之间 RRAS 和 RRAS2 蛋白丰度的箱线图。 (F) 热图显示 RRAS 低组和 RRAS 高组之间 ssGSEA 通路得分的相对丰度。(G) GSEA 显示,与 RRAS 低表达组相比,RRAS 高表达组中 MAPK 信号、RHO GTPase 循环和 ECM 通路富集。 (H) 显示 CRC 患者对西妥昔单抗治疗耐药的潜在机制的示意图。(I) RRAS 或 RRAS2 与参与三个途径的蛋白质的相关性。(J-K) 基于蛋白质丰度的CPTAC 队列中总生存期(OS) 的风险比 (HR) 和 Kaplan-Meier 曲线。 (L) 蛋白质在血浆发现队列中预测药物敏感性的 ROC 曲线及其在组织和血浆验证队列中的预测性能。
05
结直肠癌多疗程西妥昔单抗治疗预测模型的构建和验证
在纵向队列中,一个子集(定义为子队列1)由22名首次治疗时对西妥昔单抗治疗敏感、在随后的7个疗程西妥昔单抗治疗中逐渐耐药的CRC患者组成,其中收集了105份血浆样本;另一个子集(定义为子队列2)由稳定敏感组(SSG:18名患者,38个样本)和稳定非敏感组(SNSG:58名患者,153个样本)组成。
为了确定多个治疗过程中与西妥昔单抗治疗反应相关的蛋白质动态变化,他们重点关注纵向队列的亚队列 1。根据西妥昔单抗治疗反应轨迹,对治疗期间对西妥昔单抗治疗敏感(S)/不敏感(NS)的CRC患者的分布进行统计分析。在西妥昔单抗治疗期间,NS的比例随着采样时间的延长而逐渐增加(图 5A)。然后,他们探索了多个治疗过程中血浆蛋白水平的调节,结果发现139个显著正相关(正相关-sig)和374个显著负相关(负相关-sig)蛋白(图 5B)。
他们还在CRC患者中发现了西妥昔单抗治疗的非敏感生物标志物(图 5C)。K-means图显示,敏感的生物标志物在西妥昔单抗治疗期间随着采样时间的增加呈现逐渐下降的趋势,而非敏感的生物标志物在西妥昔单抗治疗期间随着疗程的增加呈现逐渐上升的趋势(图 5C-D)。他们观察到这些生物标志物在SSG和SNSG中存在明显的差异表达,以及西妥昔单抗治疗七个疗程的动态变化(图 5D)。这凸显了血浆生物标志物在西妥昔单抗连续多个疗程期间有效监测的潜在应用。
在确定了与西妥昔单抗治疗反应相关的蛋白质水平波动的生物标志物后,他们接下来着手确定这些生物标志物是否可用于预测 CRC 患者在连续疗程期间对西妥昔单抗治疗的反应。采用对噪声和过拟合具有鲁棒性的逐步逻辑回归来识别能够准确区分 SSG 和 SNSG(称为 S-sig 和 NS-sig)的特征子集。S-sig 包括 IDH3G、MDN1 和 KLC4,而 NS-sig 包括 MYL9、SBF1 和 HTRA3。为了训练和随后测试模型,根据样本类型对样本进行分区;其中,60%的样本作为训练集,剩余的40%代表独立测试集。基于S-sig和NS-sig,他们对训练集进行10倍交叉验证,得到了高AUC(0.756)的预测模型;当应用于测试集时,预测模型的 AUC 也达到了 0.797(图 5E)。这表明 S-sig 和 NS-sig 蛋白(IDH3G、MDN1、KLC4、MYL9、SBF1 和 HTRA3)的组合可以预测 CRC 患者对西妥昔单抗治疗的反应。将预测模型应用于不同的治疗过程后,他们观察到预测模型在整个治疗过程中表现良好,准确度为0.724。条形图显示了该血浆蛋白组在不同治疗过程的子集中识别敏感组和非敏感组的能力(图 5F)。重要的是,该模型在预测第二个疗程(0.818)、第三个疗程(0.737)和第四个疗程(0.800)的治疗效果方面也取得了很高的准确性(图 5F)。
他们在西妥昔单抗治疗的多个疗程中观察到这些特征蛋白的一致表达模式,表明这些生物标志物在预测个体患者的西妥昔单抗反应方面的有效性。随后,预测模型的准确性在由 31 名 CRC 患者、77 个血浆样本组成的独立队列中得到进一步验证,该队列涵盖两个疗程的西妥昔单抗治疗。在血浆验证队列中,混淆矩阵分析的结果显示,第一个疗程的准确度为 0.929,第二个疗程的准确度为 1(图5G)。综合来看,该预测模型在不同治疗过程中均具有良好的预测性能,并在独立队列中得到了进一步验证。
他们进一步评估了在七个疗程治疗的一个病例中应用的预测模型的性能。在典型病例中,发现模型中NS-sig蛋白表达量与治疗期间采样时间呈极显著正相关;而模型中S-sig蛋白表达量与治疗期间采样时间呈极显著负相关,这与治疗反应评估相关的MRI图像一致(图 5H-I) 。这些结果证明了该预测模型中包含的蛋白质的动态变化与临床治疗反应之间的一致性,这进一步验证了预测模型组的稳定性。综上所述,本项研究建立了预测模型,可应用于CRC的连续多疗程西妥昔单抗治疗,并在监测治疗过程中取得了良好的效果。
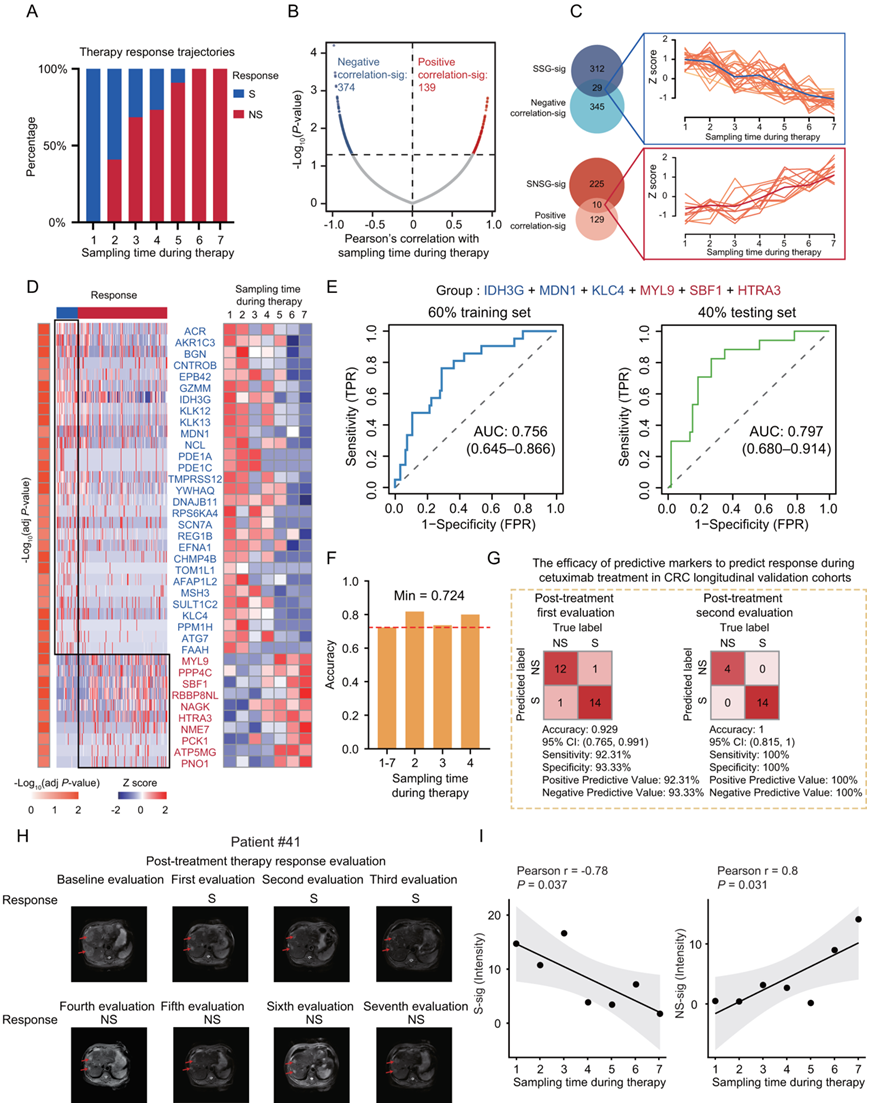
图5. CRC 连续多个疗程西妥昔单抗治疗反应预测模型的构建。
(A) 西妥昔单抗治疗期间敏感(S)/不敏感(NS)CRC患者的分布。(B) 西妥昔单抗治疗期间蛋白质动态变化丰度与采样时间的相关性。 (C) 上:维恩图显示负相关标志和ssg标志蛋白重叠。下:维恩图显示了正相关标志和SNSG标志蛋白的重叠。(D) 热图显示了这些特征蛋白在 SSG 和 SNSG 中的差异表达。 (E) 稳定反应队列中 60% 训练集和 40% 测试集中一组特征蛋白预测西妥昔单抗治疗反应的 ROC 曲线。(F) 预测模型在不同采样时间预测西妥昔单抗治疗反应的准确性。(G) 使用逻辑回归分类器区分血浆纵向验证队列中的 S 和 NS 的分类误差矩阵。 (H) 一个典型病例在整个治疗过程中每个采样点的MRI图像评价。(I) 预测模型中特征蛋白面板蛋白表达与疗程的相关性分析。
+ + + + + + + + + + +
结 论
本项研究对来自 147 名接受多疗程西妥昔单抗治疗的 CRC 患者 (CRC) 和 90 名健康对照 (HC) 的 641 份血浆样本进行了纵向和深度蛋白质组学分析。将COL12A1、THBS2、S100A8 和 S100A9 鉴定为潜在蛋白质,可在血浆和组织验证队列中区分 CRC 和 HC。本项研究确定了用于初始反应预测的潜在生物标志物(RRAS2、MMP8、FBLN1、RPTOR 和 IMPDH2)。在纵向设置中,识别出具有明显波动的两个聚类,并以高精度构建模型来预测纵向响应,并在独立队列中进一步得到验证。这项研究揭示了肿瘤诊断的不同生物标志物的异质性、第一个疗程和多疗程西妥昔单抗治疗中的初始和纵向反应预测,最终可能有助于 CRC 的监测和干预策略。
+ + + + +



