

 English
English文献解读|Nature(64.8):肺腺癌上皮细胞状态和可塑性图谱
✦ +
+
论文ID
原名:An atlas of epithelial cell states and plasticity in lung adenocarcinoma
译名:肺腺癌上皮细胞状态和可塑性图谱
期刊:Nature
影响因子:64.8
发表时间:2024.02.28
DOI号:10.1038/s41586-024-07113-9
背 景
由于筛查的加强,早期肺腺癌 (LUAD)越来越多地在早期病理阶段发现。然而,患者预后仍然是中等到较差的,因此需要改进早期治疗策略。解码驱动LUAD的最早事件可以确定调控的理想靶标。先前的研究表明,吸烟导致普遍的分子(例如KRAS突变)和免疫变化,这些变化在LUAD及其相邻的正常生态系统之间共存,并且与肺恶性病变和LUAD的发展密切相关。然而,这些报告大多侧重于肿瘤和肺正常组织的远处部位。因此,扩大LUAD景观的细胞和转录表型仍未得到充分研究。此外,尽管许多肺单细胞转录组分析(scRNA-seq)研究已经分析了免疫和基质状态,但对上皮细胞知之甚少。
实验设计

结 果
01

上皮细胞转录图谱
本项研究结合了早期 LUAD 临床标本的深入 scRNA-seq 以及接触烟草致癌物后 LUAD 发展人类相关模型中的跨物种分析和谱系追踪(图1a)。研究者团队使用 scRNA-seq 研究了来自 16 名患者的早期 LUAD 样本和 47 对正常肺(NL)样本的 EPCAM 富集上皮细胞亚群,这些样本涵盖了 LUAD 的拓扑连续体,即肿瘤相邻、肿瘤中间和肿瘤远处位置(图1a)。他们还从相同区域收集了肿瘤和正常组织集,进行全外显子组测序 (WES) 和高分辨率空间转录组学 (ST) 和蛋白质分析(图1a)。通过整合推断的拷贝数变异(inferCNV)、聚类分布、谱系特异性基因表达和具有KRASG12D体细胞突变的信息 ,将恶性细胞与其他非恶性正常细胞区分开来(图1b-c)。对非恶性聚类的分析确定了两个主要的谱系,肺泡和气道以及一小部分增殖细胞。气道细胞包括基底细胞(KRT17+)、纤毛细胞(FOXJ1+)、棒状细胞和分泌细胞(SCGB1A1+),以及罕见的细胞类型,如离子细胞(ASCL3+)、神经内分泌细胞(ASCL1+)和簇状细胞(GNAT3+)。肺泡细胞由肺泡1型(AT1)细胞(AGER1+ETV5+)、AT2细胞(SFTPB+SFTPC+)、SCGB1A1+SFTPC+双阳性细胞和一群肺泡中间细胞(AIC)组成,这些细胞紧密地夹在AT1和AT2细胞群之间,与两种主要肺泡细胞类型共有基因表达特征(图1b)。恶性细胞表现出谱系特异性标记的低表达或不表达,总体而言,谱系特征减少(图1b)。恶性细胞形成14个聚类(图1c),主要是患者特异性的,这表明患者之间存在很强的异质性。总体而言,恶性细胞表现出高水平的非整倍体。基于基因组图谱(WES)的注释显示,3例KRAS突变LUAD患者的恶性细胞(KM-LUAD)(患者P2、P10和P14)紧密聚集在一起。相比之下,来自其他luad的恶性细胞表现出更分散的聚集模式(图1d)。scRNA-seq分析证实,在患者特异性肿瘤集群中存在拷贝数变异(cnv)和KRASG12D突变,而KRAS野生型LUAD (KW-LUAD)中不存在KRASG12D。
KM-LUAD 中的恶性细胞聚集在一起,与EGFR突变体 LUAD(EM-LUAD)或MET突变体 LUAD(MM-LUAD)的细胞有明显区别(图1e)。与其他 LUAD 相比,KM-LUAD 在样本和细胞水平上均表现出更高的转录组相似性。大多数KRAS突变恶性细胞与其他细胞分开聚集,这表明KRAS突变细胞中存在不同的转录程序(图1f)。恶性细胞的分化状态表现出高度的患者间异质性。也就是说,无论肿瘤突变负荷如何,KM-LUAD 细胞分化程度最低,其次是 EM-LUAD(图1g-h)。分化状态存在肿瘤内异质性 (ITH)(例如,患者 P2、P9、P14 和 P15),14 名可检测到恶性细胞的患者中有 7 名的恶性细胞表现出广泛的 CytoTRACE 评分分布,其中 KM-LUAD 显示出比 EM-LUAD 或其他 LUAD 更高的分化变异性趋势(图1h)。

图1. 早期 LUAD 中肺上皮细胞和恶性细胞的转录景观。
(a) 实验设计和分析工作流程的示意图。(b) 十个正常上皮细胞和一个恶性细胞亚群的选定标记基因的比例和平均表达水平。(c) 17064 个恶性细胞的无监督聚类。(d) 恶性细胞的均匀流形近似和投影 (UMAP) 。(e) 恶性细胞的主成分分析 (PCA) 图。(f) 恶性细胞的 UMAP 图。(g) 由 CytoTRACE 推断的恶性细胞分化状态的 UMAP ,不同驱动突变样本的恶性细胞之间的 CytoTRACE 评分比较。(h) 每个样本的恶性细胞 CytoTRACE 评分分布。
根据 23 个复发性元程序 (MP) 的水平对恶性细胞进行聚类(Meta C1-Meta C5),结果表明 Meta C1 主要由来自 KM-LUAD 的细胞组成 (92%)。Meta C1 中的细胞还显示出与胰腺导管腺癌 (MP30)、上皮间质转化 (EMT-III;MP14) 和上皮衰老 (MP19) 中存在的KRAS G12D相关的基因模块的最高表达,相反,肺泡 MP (MP31) 的水平最低(图S2a-c)。值得注意的是,KM-LUAD患者P2、P10和P14的恶性细胞中MP30的表达明显高于KW-LUAD患者(图S2d)。恶性细胞状态在KM-LUAD中也表现出ITH(如患者P14)(图S2e)。KRASG12D细胞的一个子集显示MP30的激活,并且在突变细胞中存在其他MP的不同激活模式(例如细胞呼吸)(图S2e-f)。总体而言,具有KRAS G12D突变的恶性细胞分化降低(图S2e),这与 KM-LUAD 中肺泡分化(MP31)的丧失一致(图S2a-b)。来自患者 P14 的恶性细胞聚类表现出不同水平的 CNV,其中富含KRAS G12D细胞的聚类包含相对较晚的 CNV 事件(例如染色体 1p 丢失、染色体 8 和 12 号染色体增益)并且肺泡特征评分降低,结果与分化减弱一致(图S2g)。根据队列中KRAS突变恶性细胞的独特表达特征(即特定于聚类 5),其与 MP30 特征强烈显著相关(图S2i)。来自 Cancer Genome Atlas (TCGA) 队列且 KRAS 特征表达相对较高的 KM-LUAD 富含活化的KRAS MP30 和 Meta C1 中增加的其他 MP(图S2j)。TCGA 中 KRAS 特征表达相对较高的 KW-LUAD 显示出明显较低的总体生存率 (OS)(图S2k)。尽管队列规模较小,但单独分析KRAS G12D突变型 LUAD时也观察到了类似的趋势(图S2k)。这些数据突出了 LUAD 细胞与转录程序之间广泛的转录组异质性,这些异质性在生物学上和可能在临床上与 KM-LUAD 相关。

图S2. LUAD 恶性细胞肿瘤间和肿瘤内异质性的特征。
(a) 基于 23 个先前定义的共识癌细胞元程序 (MP) 的表达,对恶性细胞进行无监督聚类。(b) 4 个代表性 MP 的特征分数在聚类中的分布。 (c) 按复发性驱动突变状态(左)和患者(右)颜色编码的细胞中聚类 (C1-C5) 的富集。(d) 计算了每位患者的恶性细胞(左)以及 KM-LUAD 与KRAS WT LUAD(KW-LUAD,右)的 MP30。(e) P14 LUAD 恶性细胞中的 ITH 分析。(f) UMAP 图显示 P14 恶性细胞,按 3 种指示 MP 的表达着色。(g) 基于推断的 CNV 谱对 P14 恶性细胞进行无监督聚类分析和轨迹分析。(h) CNV 聚类中的肺泡 MP 表达。(i) UMAP 图(左)。KM-LUAD 恶性细胞中 MP30 表达与 KRAS 特征评分之间的相关性(右)。(j) 热图显示 TCGA LUAD 样本中所示 MP 和特征的分数分布。(k)生存分析。
02
LUAD 中的 AIC
与多区域NL样本相比,AT2细胞的LUAD总体下降,而AIC则呈现相反的模式。AIC 处于 AT2 到 AT1 细胞发育和分化轨迹的中间位置(图2a)。LUAD 组织中分化最低的 AIC 比例高于分化较高的 AIC(分别为 29% 和 11%)。值得注意的是,他们推断AIC会转变为恶性细胞,包括相对于EGFR突变恶性细胞更晚发育的KRAS突变细胞(图2a)。对AIC的进一步分析发现了KRT8明显高表达的亚群(图2b)。这些KAC增加了CDKN1A、CDKN2A、PLAUR和肿瘤标志物CLDN4的表达(图2b)。与其他AIC相比,KAC分化程度也显著较低,且发育较晚(图2c)。值得注意的是,KAC在伪时间内转化为KRAS突变的恶性细胞,而其他AIC与向AT1细胞的分化更密切相关。相对于多区域NL组织,在非恶性上皮细胞中,KAC的比例在LUAD中显著且强烈地增加(图2d),并且在LUAD中显著高于AT1、AT2或其他AIC分数(图2e)。值得注意的是,与NL来源的KAC相比,肿瘤相关的KAC聚集在离AIC更远的地方。
对KRT8、CLDN4和泛细胞角蛋白(PanCK)的高分辨率、多重成像分析表明,KAC富集于肿瘤邻近正常区域(TAN),并且与KRT8和CLDN4高表达的恶性细胞紧邻(图2f)。虽然KAC也在未受累的NL样本中发现,这与scRNA-seq分析一致,但只有在TAN中它们显示出“反应性”上皮细胞的特征(图2f,图S4a)。患者P14肿瘤组织的ST分析显示,KRT8在肿瘤区域(具有高CNV评分)和TAN区域的表达增加,这些区域在组织学上由高活性肺细胞组成,并表现出中低CNV评分(图2g)。反卷积显示,相对于肺泡细胞,KAC更接近肿瘤区域(图S4b)。肿瘤区域的NKX2-1表达和肺泡特征显著减少(图S4b),这一结果与KM-LUAD中肺泡分化减少一致。
KAC标记物在肿瘤区域和反应性肺细胞的TAN中含量高,并且它们在空间上与KRAS特征重叠(图2g)。与KRAS相似,但与AT1和肺泡特征不同,KAC特征相对于AT1、AT2或其他AIC而言是KAC中最高的(图2h,图S4c-d)。与其他AIC相关的特征在KAC中明显低于其他AIC(图S4e)。在所有样本的KAC中,KAC和KRAS特征呈正相关,与对应的肺泡上皮特征呈负相关(图2h)。相反,“其他AIC”与KRAS或肺泡特征之间没有相关性(图S4f-g)。KAC标记在KM-LUAD和恶性细胞中的表达明显高于EM-LUAD(图S4h)。与“其他AIC”和肺泡特征相比,与匹配的未受累的NL样本相比,KAC特征在TCGA肺腺癌中显著富集。值得注意的是,随着NL、癌前非典型腺瘤性增生(AAH)和侵袭性LUAD的匹配,KAC特征显著且逐渐增加(图2i),而“其他AIC”特征则没有这种模式。
KRASG12D存在于恶性细胞中,KM-LUAD的变异等位基因频率(VAF)为78%(图2j)。KRASG12D KAC只在KM-LUAD的组织(原发肿瘤)中发现,因此,KM-LUAD的KAC中KRASG12D VAF(10%)高于所有LUAD(5%)和样本(3%)中的KAC。
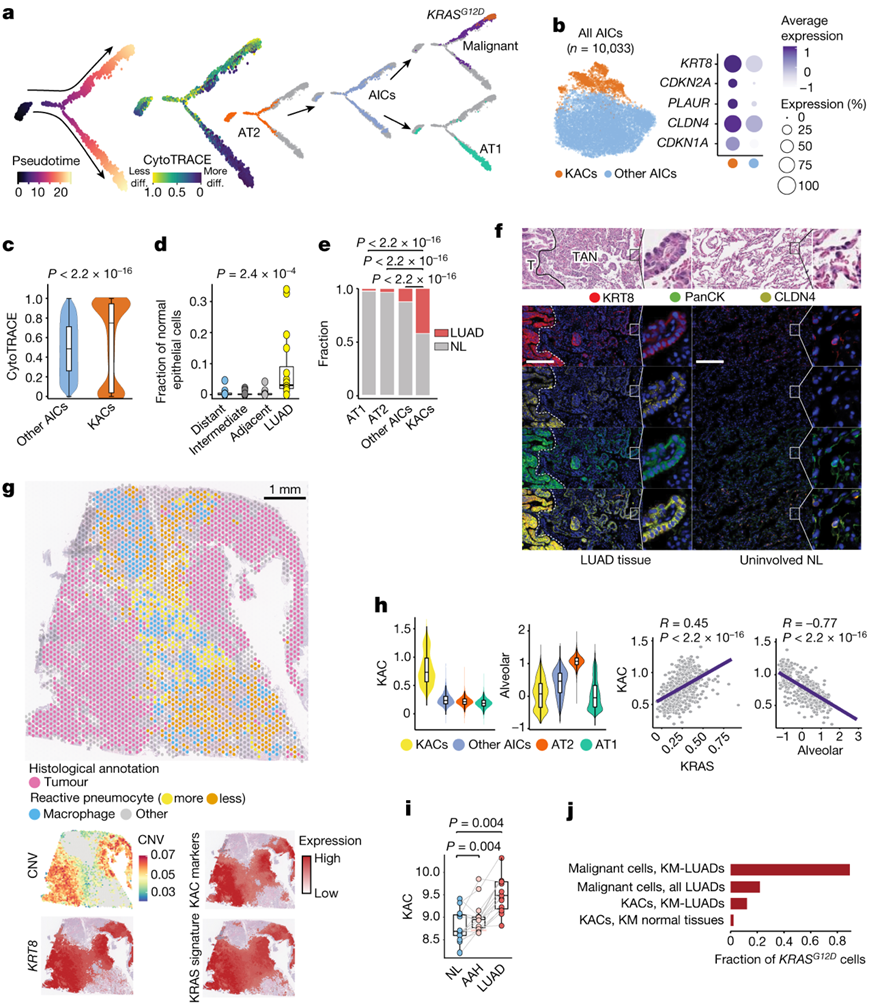
图2. 人类 LUAD 中 KAC 的鉴定和特性。
(a) 肺泡和恶性细胞的伪时间分析。(b) AIC 的亚聚类分析。(c) KAC 与其他 AIC 的 CytoTRACE 评分。(d) 非恶性上皮细胞中 KAC 的比例。(e) 按样本类型着色的肺泡细胞亚群分数。(f) H&E染色和数字空间分析显示 KRT8、PanCK、CLDN4、Syto13 蓝色核染色和合成图像。(g) 患者 P14 的 LUAD 的 ST 分析显示组织学注释的 H&E 染色的 Visium 载玻片(左)和空间热图(右)。(h) KAC、KRAS 和肺泡特征的表达(上)和相关性(下)分析。(i) 癌前病变队列中的 KAC 特征表达。(j) 不同亚群中KRASG12D细胞的比例。
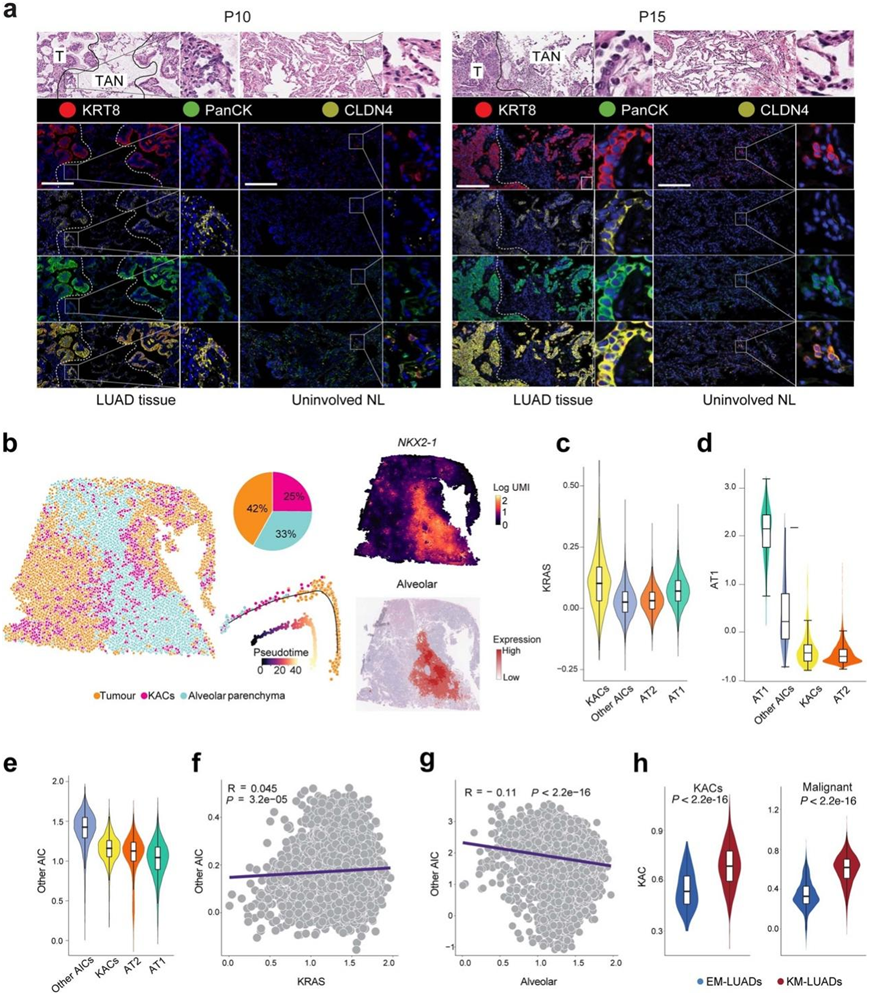
图S4.人类 KAC 的空间和分子属性。
(a) P10(左)和 P15(右)LUAD 和配对未受累 NL 组织的显微照片。(b) P14 LUAD ST 数据的 CytoSPACE 反卷积和轨迹分析。(c-e) AT1、AT2、KAC 和其他 AIC 中KRAS、AT1和其他 AIC特征的表达。(f-g) 其他AIC与KRAS 或肺泡特征评分的相关性分析。 (h) KM-或EM-LUAD样本中KAC(左)和恶性细胞(右)中KAC特征的富集。
03
KAC 状态与小鼠 KM-LUAD 相关
他们接下来对来自小鼠的肺上皮细胞进行了scRNA-seq,这些小鼠中的肺系谱特异性G蛋白偶联受体基因Gprc5a敲除(Gprc5a−/−),并且在烟草致癌物暴露后会发展成KM-LUAD(肺腺癌)。他们分析了用尼古丁衍生的亚硝胺酮(NNK)或生理盐水(作为对照)处理的Gprc5a−/−小鼠的肺部,分析时间点为暴露结束(EOE)和暴露后7个月(KM-LUAD发病时间点)(图3a)。对9272个高质量上皮细胞的聚类分析揭示了不同的细胞系,包括聚集在AT1和AT2细胞亚群之间并靠近肿瘤细胞的KAC(图S7a)。与人类细胞相似,恶性细胞表现出谱系特异性基因的低表达(图S7b)。与此一致的是,来自恶性肿瘤集群的细胞具有较高的CNV评分,表达KrasG12D突变,并表现出与肺泡分化丧失(Kng2和Meg3)和免疫抑制(Cd24a)相关的标志物的表达增加(图S7c-d)。恶性细胞仅在NNK处理后7个月出现,在致癌物和对照处理动物的EOE中不存在(图3b,图S7e-f)。与对照组相比,KAC分数在EOE时显著增加,并且在NNK处理后7个月时基本保持不变(图3b,图S7f-g)。免疫荧光(IF)分析显示,KRT8+ AT2来源的细胞存在于NNK暴露的NL中,而在盐处理小鼠的肺部几乎不存在(图3c)。LUAD 也显示出 KRT8 的高表达(图3c)。KAC 显示出KRASG12D突变的患病率明显增加,高于 CNV 负担,并且与肺泡分化丧失相关的基因表达(例如, Gnk2)增加,但与恶性细胞相比程度较小(图3d,图S7h)。值得注意的是,AT2细胞分数随着时间的推移而减少(图S7f-g)。NNK处理后7个月的ST分析显示,肿瘤区域Krt8和Plaur的表达显著增加,KAC和KRAS特征在空间上重叠(图3e,图8a-e)。与公开的人类数据一致,Krt8highKAC 具有增加的 KAC 和 KRAS 特征表达,在肿瘤周围的“反应性”非肿瘤区域中富集,并且它们本身是从正常细胞到肿瘤细胞的转变的中间体

图3. 在烟草相关的 KM-LUAD 发病机制中,KAC 在肿瘤发作早期和肿瘤发作之前进化。
(a) 体内实验设计示意图。(b) 不同治疗组和时间点的恶性细胞(左)和 KAC(右)的比例。(c) 小鼠肺组织中 KRT8、LAMP3 和 PDPN 的 IF 分析。(d) 肺泡细胞和恶性细胞之间的 CNV 评分分布和突变细胞比例。 (e) 暴露于 NNK 7 个月后肺组织的 ST 分析。

图S7. KM-LUAD 烟草致癌小鼠模型中上皮亚群的 scRNA-seq 分析。
(a) 小鼠上皮细胞亚群的 UMAP 分布。(b) 小鼠正常上皮细胞谱系和恶性细胞聚类的选择标记基因的比例和平均表达水平。(c) 肺泡和恶性细胞的 UMAP 图。(d) UMAP(上)和小提琴(下)图显示恶性和肺泡亚群中Cd24a的表达水平。(e) 按细胞谱系、KrasG12D突变状态和 CNV 评分着色的肺泡和恶性细胞的 UMAP 分布。(f) 每个样本中正常上皮细胞谱系和恶性细胞的比例。(g) 在 EOE 和用 NNK 或盐水治疗后 7 个月之间恶性细胞、KAC、AT2 和 AT1 细胞的分数变化。(h) UMAP(上)和小提琴(下)图显示恶性和肺泡细胞亚群中Gkn2的表达水平。
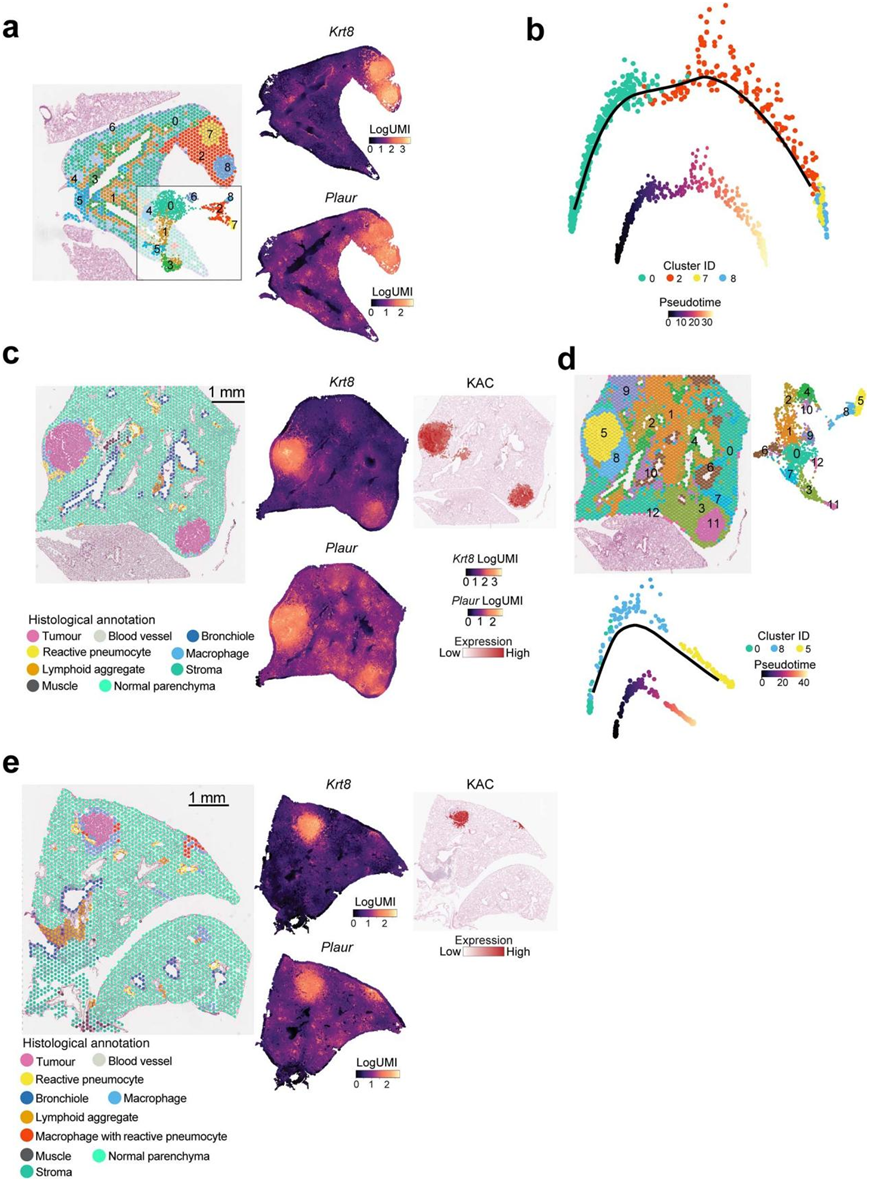
图S8. 与烟草相关的 KM-LUAD 发展中的 KAC 的 ST 分析。
(a) 荷瘤小鼠肺的 ST 分析,其中 Seurat 识别的细胞聚类并进行空间映射。右侧显示了具有Krt8和Plaur表达的空间图。(b) 同一荷瘤小鼠肺的 C0(肺泡实质)、C2(肿瘤附近有 KAC 的反应区)以及聚类 C7 和C8(代表两个肿瘤)的伪时间轨迹分析。(c) (a) 中相同的 NNK 暴露小鼠的另一个荷瘤肺区域进行 ST 分析。(d) 伪时间轨迹分析。(e) NNK 后 7 个月另一只小鼠的荷瘤肺的 ST 分析。
04
KAC 与 AT2 向Kras突变肿瘤细胞的转变有关
小鼠和人类的KAC(肺腺癌细胞)显示了共同的通路激活增加,包括NF-κB、缺氧和p53信号等(图S9a-b)。p53特征在暴露结束时的KAC中显著增加,在NNK暴露7个月后更是如此,与AT2细胞和肿瘤细胞相比(图S9c)。类似的模式在p53途径相关基因和衰老标志物的表达中也有所体现,包括Cdkn1a、Cdkn2b和Bax,以及Trp53本身(图S9c)。值得注意的是,Krt8+过渡细胞在博来霉素诱导的肺泡再生过程中激活了p53,这些细胞显示与本项研究中的KAC有重叠基因(图S9d)。
小鼠KAC特征在小鼠KAC和恶性细胞和人类LUAD中显著富集,在癌前AAH中也显著增加,并且相对于匹配的NL组织,在侵袭性LUAD中进一步增加(图S9e-f)。与急性肺损伤中的肺泡中间产物和人类LUAD中的KAC相似,小鼠KAC很可能是AT2细胞衍生的,在AT2向AT1细胞分化中充当中间状态,并推断为向恶性细胞转化(图4a)。与其他肺泡亚群相比,KAC处于一种更接近恶性细胞的中间分化状态。与暴露后7个月相比,EOE时的KAC分化较小(图4a)。
他们使用Gprc5a−/−小鼠和报告标记的AT2细胞(Gprc5a−/−;SftpccreER/+;RosaSun1GFP/+)(图4 b)研究了 KAC 的生物学特性。GFP+类器官来自NNK暴露的报告小鼠,在EOE富集KAC。与盐处理小鼠相比,NNK处理小鼠的GFP +AT1细胞、KAC和肿瘤细胞的比例明显增加(图4c)。为了进一步证实KAC可以产生肿瘤细胞,他们在Gprc5a−/−; KRT8-creER;RosatdT/+小鼠中标记了KRT8+细胞。与生理盐水处理的对照小鼠相比,在EOE到NNK时,肺实质中tdT+标记增加(即KAC数量增加)(图4d)。值得注意的是,大多数肿瘤显示不同程度的tdT + KRT8 +细胞,一些肿瘤显示出强程度的 tdT+标记,这表明 KRT8+细胞的致癌作用(图4d)。大多数tdT+肿瘤细胞是AT2细胞衍生的(LAMP3)(图4e)。在 EOE 和 EOE 至 NNK 后的随访中, tdT + LAMP3 +细胞占总 tdT +细胞的比例相似(图4e)。正常外观区域也显示 tdT + AT1 细胞(NKX2-1+ LAMP3−),这表明 AT2 细胞和 KAC 可能转换为 AT1 细胞。
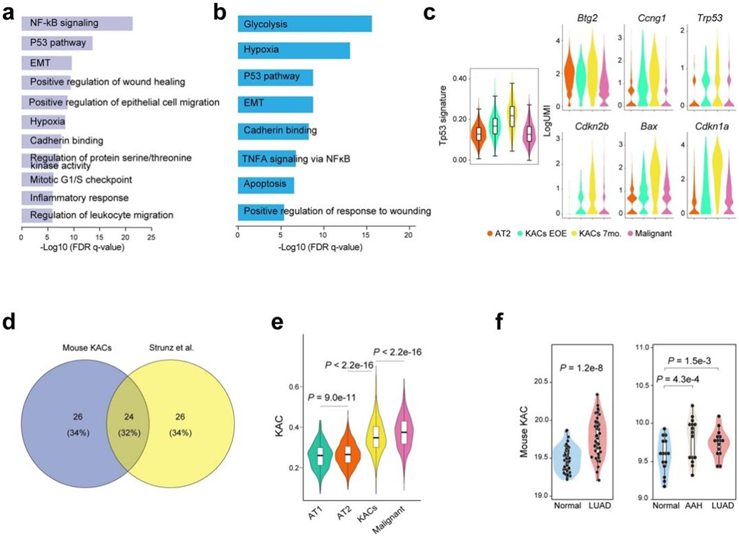
图S9. 小鼠 KAC 特征和通路与损伤模型和人类 KM-LUAD 相关。
(a-b)通路富集分析。(c)特征基因表达分析。(d) 独特和重叠差异表达基因(DEG) 集的百分比。(e-f)KAC的表达特征。
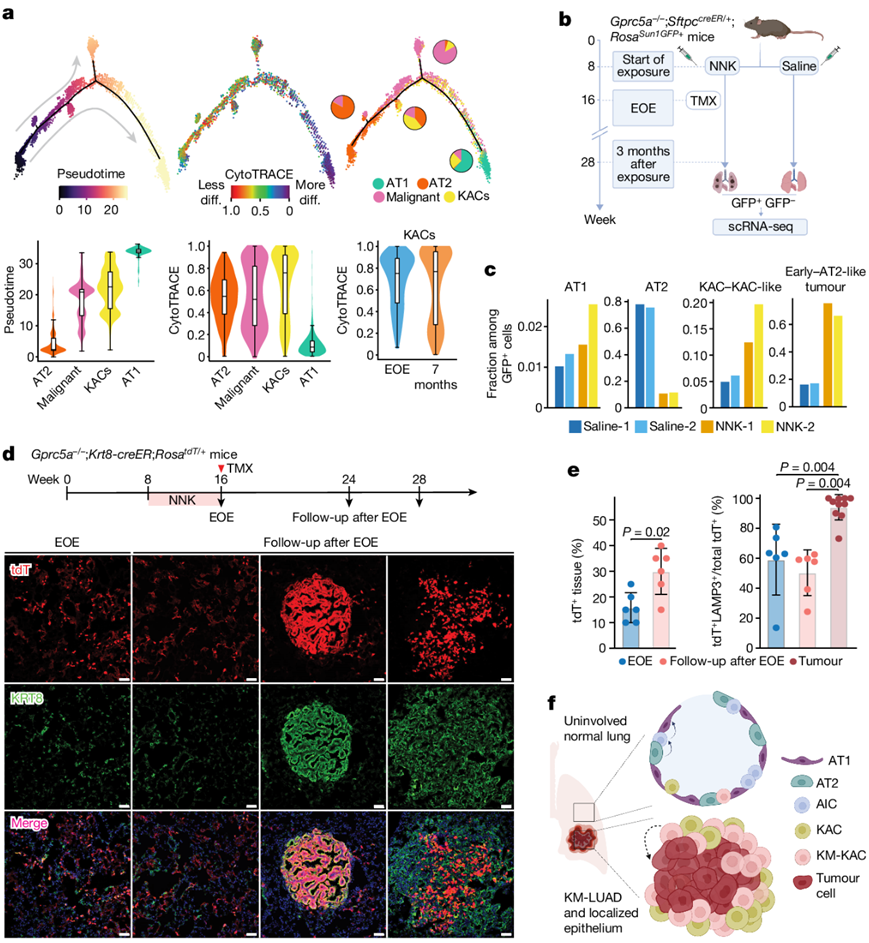
图4. KAC 与 AT2 向Kras突变肿瘤细胞的转变有关。
(a) 肺泡细胞和恶性细胞的轨迹分析。(b) 动物模型分析。(c) 在暴露后3个月,分析了来自2只nnk处理和2只盐水处理小鼠肺的GFP+细胞中的AT1、AT2、KAC和KAC样细胞和早期肿瘤和AT2样肿瘤细胞的比例。(d)免疫荧光分析。(e)(左)包含 tdT+细胞的肺组织区域百分比。(右)不同时间点正常外观区域中tdT+ LAMP3+细胞占总 tdT+细胞的百分比。(f) 建议的肺泡可塑性模型。
+ + + + + + + + + + +
结 论
本项研究了来自 16 个早期 LUAD 和 47 个匹配的正常肺样本的单个上皮细胞,包含多种正常和癌细胞状态,癌细胞之间的多样性与 LUAD 特异性致癌驱动因素密切相关。KRAS突变癌细胞表现出不同的转录特征、分化降低和低水平的非整倍性。人类 LUAD 样本周围的非恶性区域富含肺泡中间细胞,这些细胞显示KRT8表达升高、分化降低、可塑性增强和驱动KRAS突变。KAC 的表达谱在肺癌前细胞和 LUAD 细胞中富集,并表明生存率较差。在暴露于烟草致癌物的小鼠中,KAC 在肺肿瘤之前出现,并在停止接触致癌物后持续存在数月。此外,它们获得了Kras突变,并在源自AT2细胞的富含 KAC 的类器官中表现出对靶向 KRAS 抑制的敏感性。最后,在暴露于致癌物后对 AT2 细胞或 KRT8+细胞进行谱系标记表明,KAC 可能是 AT2 细胞向肿瘤细胞转化的中间体。这项研究为 LUAD 发展根源的上皮细胞状态提供了新的见解,这些状态可能包含预防或干预的潜在目标。
+ + + + +



