

 English
English文献解读|Nat Commun(16.6):抑制基质 I 类 HDAC 可抑制胰腺癌进展
✦ +
+
论文ID
原名:Inhibiting stromal Class I HDACs curbs pancreatic cancer progression
译名:抑制基质 I 类 HDAC 可抑制胰腺癌进展
期刊:Nature Communications
影响因子:16.6
发表时间:2023.12.06
DOI号:10.1038/s41467-023-42178-6
背 景
胰腺导管腺癌(PDAC)的生存率很低,预计到 2030 年将成为第二大致命癌症。预后不良和治疗抵抗部分归因于转化胰腺上皮诱导的促结缔组织增生反应导致的突出的活化间质。结缔组织增生反应主要是由于成纤维细胞样细胞的激活,包括胰腺星状细胞(PSC),这是胰腺基质中主要的成纤维细胞。潜在的PSC激活是由静止相关的转录程序(如脂肪生成程序)转换为促结缔组织增生的转录程序,包括驱动肌成纤维细胞转分化和增殖的转录程序。
目前缺少对PDAC表观遗传机制的研究。
实验设计
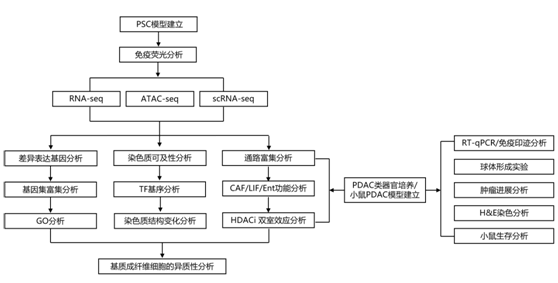
结 果
01

HDAC通过诱导血清反应因子(SRF)/ FOXM1诱导的促纤维原增生转录程序促进PSC的激活
为了测试组蛋白去乙酰化酶(HDAC)抑制(HDACi)调节PDAC中基质激活的潜力,研究者团队首先确定了entinostat (Ent) (III期研究中的I类HDAC抑制剂)在体外调节PSC激活的能力。在没有任何药物处理的情况下,从正常小鼠胰腺中培养新鲜分离的PSCs, 3-6天诱导激活表型,其特征是失去静止特异性细胞质脂滴,诱导肌成纤维细胞标记α-SMA (α-平滑肌肌动蛋白,基因名称为Acta2),以及诱导增殖标记Ki67(基因名称为Mki67)的表达和结合EdU的能力(图1b-c)。此外,多种肌成纤维细胞和增殖基因,作为促结缔组织增生转录程序的一部分,在PSC激活过程中发生显著诱导。尽管HDAC通常是转录抑制因子(因为它们具有去除激活转录的组蛋白乙酰化的能力),但从第1天开始用Ent处理PSC,在培养诱导的激活过程中,会阻断肌成纤维细胞和增殖基因的诱导,包括α-SMA和Ki67(图1b-c)。与增殖标志物的表达降低一致,在培养诱导的活化过程中,Ent处理抑制EdU标记并以浓度依赖的方式减少PSC细胞数量(图1b-c)。
为了分析HDAC调控的转录程序,他们比较了在存在和不存在Ent的情况下,PSC体外激活诱导的全基因组表达变化。PSC激活的转录变化受到Ent的广泛抑制,包括全面抑制促结缔组织增生前转录程序,这些转录程序控制着增殖的重要基因,以及那些编码细胞外基质(ECM)、细胞骨架、粘附和肌成纤维细胞典型的信号成分的基因(图1d-e)。基因集富集分析(GSEA)证实,体外激活诱导的基因集的富集包括Ent处理下调的基因集(图1f),在体外PSC激活期间诱导的基因中,大约有一半由Ent抑制而发生显著减少(图1g)。GO分析显示,这些基因在功能上富集于与PSC激活相关的通路,包括增殖和肌成纤维细胞特性相关通路,突出了HDACi抑制驱动PSC激活的功能转录程序的可能性(图1h)。约30%的PSC激活下调基因的抑制由Ent逆转(图1i)。有趣的是,脂质代谢相关基因的去抑制,包括Fabp4(脂肪酸相关蛋白4)(图1d-j),静止PSC和正常基质的标记,表明Ent至少保留了部分静止相关的脂肪生成程序。
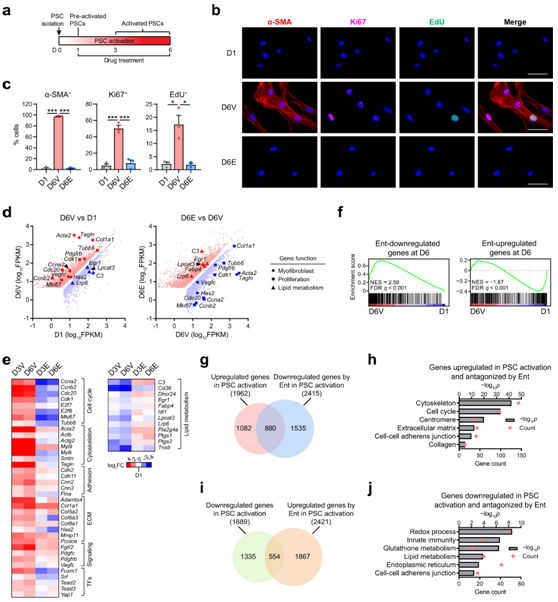
图1. HDACi 通过转录调控抑制 PSC 激活。
(a) 药物治疗体外 PSC 激活方案。 (b-c) 免疫荧光染色的代表性图像和定量。 (d) RNA-seq数据的散点图。(e) 热图显示了在有或没有 Ent 的情况下 PSC 激活中选定功能基因的 PSC 样品中的表达差异倍数 (FC)。(f) GSEA 图显示 D6 时前 500 个 Ent 下调和上调基因分别位于 PSC 激活中诱导和抑制的基因中。(g-h) 维恩图比较了在 D6 时 PSC 激活上调的基因和 Ent 下调的基因,以及GO分析。 (i-j) 维恩图比较了 PSC 激活中下调的基因和 D6 处 Ent 上调的基因以及 GO 分析。
02
SRF-FOXM1 TF轴在PSC激活中介导HDAC协调的转录程序
接下来,他们分析了公开数据库,以确定可以优先结合由体外激活诱导并由Ent拮抗的基因子集的转录因子(TF)或辅因子(图1g)。根据肌成纤维细胞的特征或相关功能对转录调节因子列表进行优先排序,确定FOXM1、SRF、YAP1和TEAD家族TF作为候选驱动因子(图2a)。这些转录调节因子的表达与其靶标的表达是同步的,通过体外激活诱导,并通过Ent处理(图1e)和HDAC抑制。
为了评估这些可能的转录驱动因子的功能,他们确定了FOXM1和SRF敲低对活化的PSC中促结丝增生前转录程序的影响(图2b)。在体外激活诱导的基因中,约10%的基因随着FOXM1的缺失而表达降低(图2c)。这些依赖FOXM1的基因在细胞周期进程的通路中富集(图2b-e),与FOXM1在调节增殖中的作用一致。SRF的缺失抑制了约25%的体外激活基因集的诱导,包括大多数FOXM1依赖的增殖基因以及对FOXM1不敏感的肌成纤维基因(图2b-g),这与SRF在诱导肌成纤维细胞激活中的作用一致。总之,这些结果表明,HDAC部分通过SRF和FOXM1的活性来协调诱导促纤维原增生转录程序。
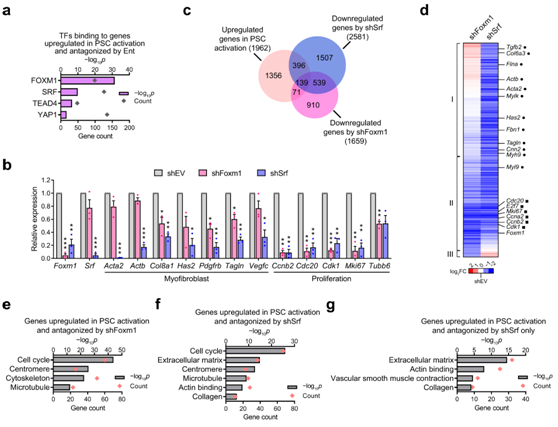
图2. SRF-FOXM1 TF 轴介导 PSC 激活中 HDAC 协调的转录程序。
(a) 在 PSC 激活中诱导并由 Ent 抑制的基因处富集 TF 结合位点。(b) RT-qPCR结果。(c) 维恩图显示经 RNA-seq 鉴定,激活的 PSC 中显著上调的基因以及 shFoxm1 或 shSrf下调的基因的分布。 (d) 层次聚类热图显示 FOXM1 和 SRF 消耗对体外激活诱导的基因和 TF 消耗抑制的基因的影响。(e-g) GO分析。
03
HDAC协调PSC激活中的染色质变化
为了深入研究HDAC如何协调PSC激活中的转录程序的机制,他们利用转座酶染色质可及性测序(ATAC-seq)进行分析,探究了染色质可及性的全基因组变化。体外激活导致可达位点增加约80%,包括基因和基因间位点增加约90%,以及启动子位点增加约15%(图3a)。值得注意的是,这些变化主要是由于新位点的增加(93%),而已有位点的缺失很小(14%)(图3b)。总的来说,超过55000个位点的可及性增加了2倍,整体可及性平均增加了4倍(图3c-f),这些结果将PSC激活与基因组中染色质可及性的增加联系起来。
GSEA显示,与这些可及位点相关的基因,以及与基因、基因间或启动子位点特异性相关的每个亚群,在PSC激活期间富集(图3g)。启动子中可及性较高的基因组位点与转录诱导程度较高相关。与此同时,对PSC激活和Ent抑制基因的基因组位点或附近基因的研究也显示,激活后可及性普遍增加,包括与肌成纤维细胞特性和增殖相关的基因亚群,以及相关的TF基因(图3h-j)。在预测的调控区域如启动子中存在新生位点(图3h),暗示在PSC激活过程中,染色质开放是诱导转录的先决条件。
鉴于Ent能够抑制转录变化,他们探索了这种能力是否由染色质可及性的变化驱动。与活化的PSC相比,在体外活化期间包含HDAC抑制剂导致可及的染色质位点减少约25%(图3a-b)。在激活的PSC中,约有30000个位点,包括约23000个激活后可及性增加的位点,在Ent处理下显示可及性降低(图3c-i)。重要的是,激活引起的可及性增加在大多数位点(93%)减少了,可及性平均减少了50%(图3d-f)。此外,激活后可及性高度增加和Ent处理后高度减少的基因组位点与肌成纤维细胞特性和增殖功能的基因相关(图31)。
综上所述,这些结果证明了HDAC能够中断原丝胞浆转录程序激活所需的染色质变化,促进HDAC在PSC激活中协调染色质结构变化的作用。
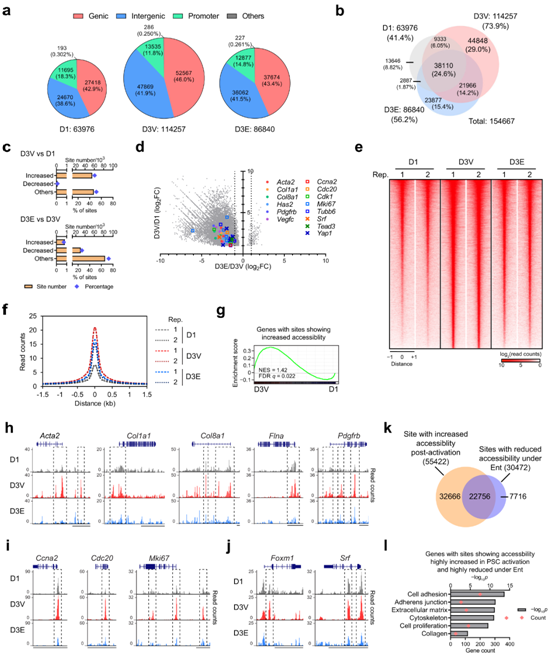
图3. Ent处理限制了PSC激活过程中染色质的打开。
(a) 通过 ATAC-seq 检测到的可及位点的数量和百分比及其基因组注释。(b) 维恩图显示 PSC 样本中可及性位点的分布以及相对于检测到的位点总数的百分比。 (c) 可及性显著增加或减少的位点数量。(d) 散点图显示基因组位点激活后(y轴)和 Ent 治疗下(x轴)的可及性变化。 (e-f) 热图显示归一化 ATAC-seq 读取计数,直方图显示基因组位点的平均归一化读取计数。 (g) GSEA 图显示激活后上调的基因在基因组位点显示可及性大大增加的基因中富集。(h-j) 基因组浏览器跟踪代表性 PSC 样本中选定的肌成纤维细胞、增殖和 TF基因位点,并突出显示突出显示差异可及性的基因组位点。(k) 维恩图显示了激活后可及性显著增加的基因组位点和 Ent 处理后可及性显著降低的基因组位点分布。(l) GO分析。
04
HDACi抑制癌症相关成纤维细胞(CAF)激活和TGF-β-和TNF-α-诱导的反应
由于来自PDAC的CAF通常携带与活化的PSC相似的表型特征,他们探索了HDACi是否能够以类似的方式调节来自小鼠PDAC模型和人类PDAC患者的CAF的转录程序。Ent处理下调了小鼠和人CAF中类似的一组肌成纤维细胞和增殖标记物,以及TF、SRF和FOXM1(图4a-c)。Ent处理还上调了小鼠和人类CAF中类似的脂质相关基因(如Fabp4/ Fabp4)(图4a-b)。在CAF中,Ent处理后下调的基因在增殖和与肌成纤维细胞功能和特性相关的生物过程中显示出功能富集,而Ent处理后上调的基因在脂质代谢和其他生物过程中富集(图4d),进一步证实了HDACi逆转导致CAF激活的转录程序开关的能力。
此外,Ent处理下调TGF-β信号通路相关基因(图4d-e),TGF-β信号通路是一种诱导肌成纤维细胞转分化的促粘连增生通路,表明HDAC在间质成纤维细胞整合环境信号中发挥关键作用。
为了探究HDAC在调节TGF-β通路中的作用,他们研究了Ent如何在患者源性CAF的转录水平上影响TGF-β反应。总的来说,Ent处理减弱了TGF-β的作用,拮抗转录激活和抑制(图4f)。值得注意的是,Ent下调了约60%的TGF-β诱导的靶标(图4g)。这组基因在功能上富集于与肌成纤维细胞特性相关的通路,包括关键标记,如α-SMA (ACTA2)和transgelin(基因名称TAGLN)(图4g-i)。
同样,Ent处理系统地下调了TNF-α信号通路相关基因(图4d-e),TNF-α信号通路是TME中活跃的旁分泌途径,能够在间质成纤维细胞中诱导炎症/促肿瘤表型。Ent处理后下调的TNF-α靶标包括白血病抑制因子(LIF)(图4a-c),LIF是一种促肿瘤细胞因子,可触发肿瘤STAT3通路,促进PDAC进展和治疗耐药,表明HDAC有可能降低CAF分泌组的致瘤性。此外,在CAF中,对TNF-α处理的总体转录变化由Ent抑制。Ent将TNF-α介导的转录激活中位数从2.3倍降低到1.5倍,而TNF-α介导的抑制从0.44倍降低到0.16倍(图4j)。有趣的是,Ent对>50%的TNF-α诱导基因集的影响优于TNF-α刺激(图4k)。Ent抑制TNF-α-诱导的基因在促炎反应相关的通路上显示功能富集,包括NF-κB和TNF-α途径的主要功能效应物,如LIF(图4k-m)。综上所述,这些数据表明,HDACi不仅可以逆转CAF中成纤维细胞激活转录程序的开关,还可以拮抗由TME信号驱动的促结缔组织增生和促炎症反应,这意味着可能降低PDAC基质中的促肿瘤作用。
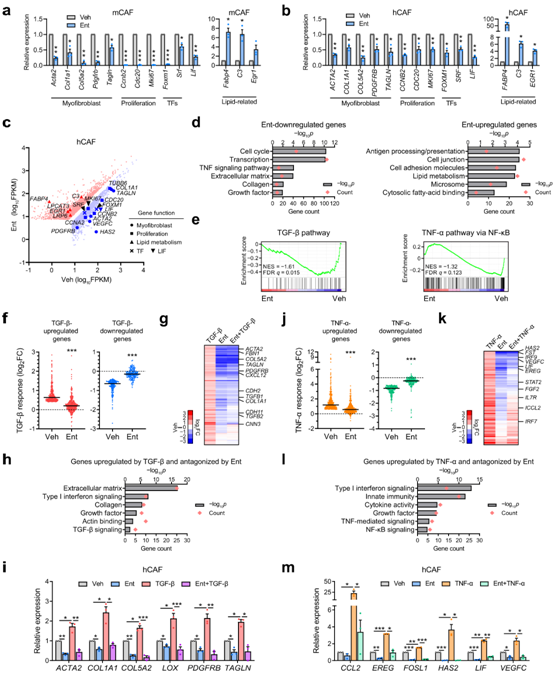
图4. Ent 抑制 CAF 激活以及 TGF-β 和 TNF-α 诱导的反应。
(a-b) RT-qPCR 数据显示小鼠 (m) (imCAF1) 和人 (h) CAF (ONO) 细胞经过 2 天Ent 处理 (10 μM) 后代表性功能基因的表达。 (c) RNA-seq 数据的散点图显示 ONO 中 Ent显著上调或下调的基因。(d)GO分析。(e) GSEA 图显示 Ent 下调基因中 TGF-β 和 TNF-α 途径成分的富集。(f) 点图显示,在载体(Veh)或 Ent 处理后,ONO 中 TGF-β 上调(333)或下调(170)基因的 TGF-β诱导表达变化。 (g) 层次聚类热图显示 TGF-β、Ent 或两者在 TGF-β 诱导且对 Ent 定向抑制敏感的基因上的表达变化。 (h)GO分析。(i) RT-qPCR 结果证实了 Ent 对 ONO 中选定的 TGF-β 诱导基因的影响。 (j) 上调或下调的基因。 (k) 热图显示 TNF-α、Ent 或两者处理下 TNF-α 上调且对 Ent 定向抑制敏感的基因的表达变化。 (l) GO分析。 (m) RT-qPCR 结果证实了 Ent 对 YAM 中选定的 TNF-α 诱导基因的影响。
05
HDACi减少了caf介导的促肿瘤信号
Ent抑制LIF表达的能力促使他们研究HDACi是否通过调节旁分泌信号来降低CAF的促瘤活性。在降低Lif/ Lif表达的同时(图5a-b),Ent处理将人和小鼠CAF培养的条件培养基(CM)中的Lif丰度降低了约75%(图5c-d)。
为了评估HDACi对CAF分泌体致瘤性的影响,从载体和Ent-处理的CAF中收集条件培养基(CM),通过离心过滤去除含有Ent的小分子部分(<3 kDa),并分析其激活致瘤性STAT3通路和促进PDAC细胞中锚定非依赖性球体形成的能力。将经Ent处理的CAF的PDAC细胞置于条件培养基(CM)中,导致磷酸化的STAT3 (pSTAT3)减少(图5e-f),这是LIFR-STAT3通路活性的标志,表明HDACi作用下来自CAF的促肿瘤信号减少。用抑制性抗LIF抗体(α-LIF)处理后,pSTAT3显著降低,这表明LIF是CAF分泌组激活STAT3的主要驱动因子(图5e-f)。一致地,来自Ent处理的CAF的CM在促进PDAC细胞的球形形成方面效果较差,然而,添加重组LIF可以恢复球状体的形成(图5h),这意味着HDAC抑制的CAF的亚水平促肿瘤潜能是由于LIF分泌不足。
与Ent处理的HDACi类似,shRNA介导的I类HDAC (HDAC1、2和3)的缺失降低了LIF的表达和分泌(图4i-j)。与此一致的是,HDAC耗竭降低了CAF分泌组激活肿瘤固有STAT3的能力(图5k)和促进PDAC细胞生长的能力(图5l)。
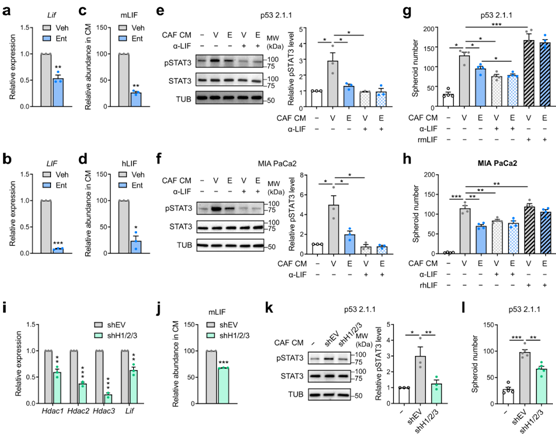
图5. HDACi 降低 CAF 介导的促肿瘤 LIF-STAT3 信号传导。
(a-b) RT-qPCR 分析。(c-d) 通过免疫测定法在经 Ent 处理的 CAF 的 CM 中检测到mLIF和 hLIF的相对丰度。 (e-f)免疫印迹分析。(g-h) 使用 CM、α-LIF 抗体 (4 μg/ml) 和/或重组 (r) m/hLIF (0.1 μg/ml) 在 p53 2.1.1和 MIA PaCa2 细胞中形成的球状体数量。 (i) RT-qPCR 分析。(j) 来自 shH1/2/3 CAF 的 CM 中 mLIF 的相对丰度。(k-l)免疫印迹和球体形成数量分析。
06
CAF中HDAC的缺失减少了肿瘤的进展
为了进一步支持hdac在体内调节CAF致瘤性中的作用,我们将小鼠PDAC细胞与HDAC缺失的CAF进行正交异性共移植,并逐步检测肿瘤负荷。与具有正常HDAC的CAF相比,HDAC缺失CAF导致肿瘤发展过程中肿瘤体积减小(图6a-b),并且在终点处肿瘤重量降低(图6c)。HDAC缺失的CAF移植物α-SMA+成纤维细胞室减少,天狼星红(SR)+胶原含量降低(图6d),这与HDAC在调节间质促纤维形成程序中的作用一致。值得注意的是,细胞角蛋白19 (CK19)+肿瘤区在HDAC缺失的CAF移植物中也减少了(图6d),这在很大程度上与α-SMA+成纤维细胞区成正比(图6e),表明HDAC缺失的CAF致瘤性作用较低。此外,在HDAC缺失的CAF移植中也检测到较低的肿瘤内LIF丰度(图6f),这与这些CAF降低肿瘤促进作用相一致。总的来说,这些发现证明HDAC在调节LIF介导的CAF致瘤性中的关键作用,部分是通过促进LIF表达和产生LIF的成纤维细胞的增殖。
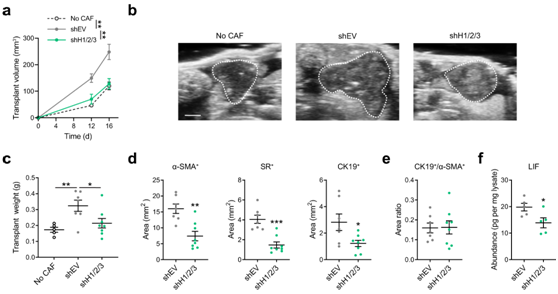
图6. CAF 中 HDAC 的消耗可减少体内肿瘤进展。
(a-b) 估计体积和原位移植超声成像。 (c) 终点处的移植重量 (D19)。(d-e) 整个移植切片中总 α-SMA、天狼星红 (SR) 和 CK19 阳性区域的测量以及 CK19 +和 α-SMA +区域的比率。(f) 通过免疫测定法测量每毫克移植裂解物的 LIF 丰度。
07
在PDAC模型中,HDACi的双室效应(Bicompartmental effects)可降低疾病严重程度
为了评估HDACi在体内的疗效,他们在KPf/fC小鼠(KrasLSL-G12D/+;Trp53f/f;Pdx1-Cre)中进行了Ent处理,这是一种PDAC以相对同步的方式进展的基因工程小鼠模型(GEMM)。KPf/fC小鼠的Ent处理显著降低了肿瘤负荷(41%)(图7a-b)并延长了生存期(35%)(图7c)。组织病理学分析显示,Ent处理减少了高级别低分化肿瘤的进展(图7d-e),暗示HDACi有能力阻止肿瘤进展。
此外,Ent处理降低了CK19+肿瘤细胞的体积,α-SMA+激活的间质成纤维细胞和SR+间质胶原含量,支持Ent双室靶向能力,以减少肿瘤进展和间质活化(图7f)。在Ent处理下也观察到较少的Ki67+增殖细胞,这与Ent在两个室中诱导细胞停滞的能力一致(图7f)。值得注意的是,肿瘤/肌成纤维细胞比例(CK19+/α-SMA+)得以维持(图7g),这与肿瘤侵袭性降低的结果表明,Ent处理避免了与间质成纤维细胞耗损相关的有害影响(图7d-f)。此外,Ent与gemcitabine (Gem)(一种经常用于PDAC患者的一线化疗药物)联合治疗,与载体治疗相比,肿瘤负担减少53%,生存期增加60%(图7b-c)。
图7. Ent 处理以阻止肿瘤进展,并为PDAC GEMM患者提供治疗益处。
(a-b) 代表性肿瘤图像和肿瘤重量测量。(c)生存曲线。(d) 肿瘤样本的病理分级。(e) 肿瘤切片的代表性图像染色。(f-g)肿瘤切片阳性区域的量化。
08
HDACi在体内改变间质成纤维细胞的异质性
随着PDAC严重程度的降低,在Ent处理下α-SMA+激活的间质成纤维细胞同时减少(图5d, 6f),暗示HDAC活性调节间质成纤维细胞异质性。为了阐明HDACi对成纤维细胞组成的影响,他们通过标记物PDPN (podoplanin)检测了从KPf/fC小鼠肿瘤分离的成纤维细胞中Ent诱导的单细胞转录组变化。
在已鉴定的10个成纤维细胞亚群中,亚群1-5高表达炎症或外层成纤维细胞的标记,如Ly6c1(Ly6C表面抗原的主要编码基因)和其他标记,但低表达肌成纤维细胞标记,如Acta2和Tagln(以下称为Ly6c1Hi亚群)(图8a-c)。相反,亚群6 - 10(称为Ly6c1Lo亚群)显示炎症/外膜标记物的低表达,但肌成纤维细胞标记物的高表达,包括典型的肌成纤维细胞亚群(亚群7-9),增殖亚群(亚群10,由Mki67标记)和具有抗原呈递潜力的亚群(亚群6,由Cd74标记)(图8a-c)。值得注意的是,Ent处理降低了两个主要的肌成纤维细胞亚群8和9的频率;同时显著富集Ly6c1Hi亚群之一的亚群3(图8d-f)。该亚群以脂质相关基因为标记(如Fabp4、Lcn2)(图8g),并显示出高表达脂肪生成转录调节因子的趋势,包括PPAR-γ(过氧化物酶体增殖物激活受体γ,基因名称Pparg)和PGC1-α (PPAR-γ共激活因子α,基因名称Ppargc1a),暗示这些成纤维细胞具有成脂潜能。
此外,Ent处理在大多数成纤维细胞亚群中上调脂肪生成标记物和下调肌成纤维细胞标记物的能力转化为在整个成纤维细胞群中可检测到的转录变化(图8h-i)。此外,在低级别高分化肿瘤中频繁检测到FABP4+脂质成纤维细胞样细胞,加上在高级别低分化肿瘤中不存在FABP4(图8j-k),将脂质成纤维细胞与良性肿瘤和活性较低的基质联系起来,这与FABP4是预测良好患者预后的顶级基质标志物之一的发现相一致。Ent处理后低级别肿瘤发生率的增加(图7d)也与Ent处理后脂肪生成成纤维细胞频率的增加(图8d-f)相一致,这意味着HDAC调节的基质转录程序的开关与肿瘤进展高度协调。总之,这些研究结果表明,HDACi处理阻断了体内脂肪生成到肌成纤维细胞的程序转换,逆转了间质激活和肿瘤进展过程中成纤维细胞组成的变化。
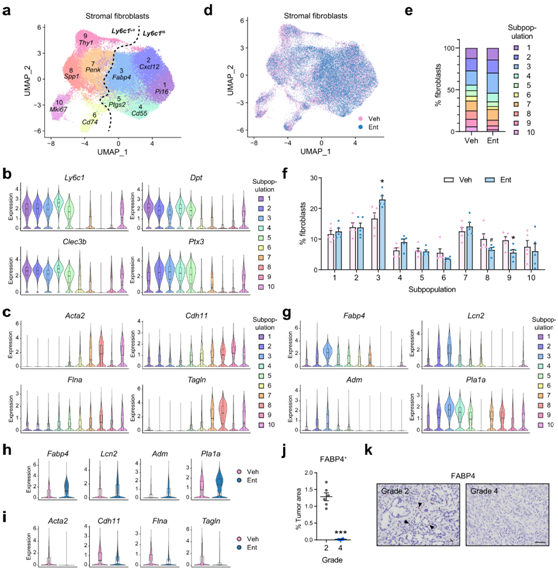
图8. Ent处理可促进脂肪生成成纤维细胞富集并降低体内肌成纤维细胞频率。
(a) UMAP显示 10 个基质成纤维细胞亚群。(b-c) 小提琴图显示成纤维细胞亚群中选定的炎症/外膜和肌成纤维细胞标记物的表达。 (d) UMAP 显示来自 Veh 或 Ent 处理的KP f/f C小鼠的基质成纤维细胞亚群的分布。 (e-f) 条形图显示成纤维细胞亚群的平均百分比。(g) 小提琴图显示脂肪生成亚群(亚群 3)的选定标记物的表达。 (h-i) 小提琴图显示 Ent 增强成纤维细胞群体中脂肪生成标记物的表达并减少肌成纤维细胞标记物的表达。 (j-k) KP f/f C小鼠2 级和 4 级肿瘤代表性区域 FABP4 染色的定量和代表性图像。
+ + + + + + + + + + +
结 论
本项研究发现HDAC是促进诱导胰腺间质成纤维细胞促结缔组织增生和促肿瘤发生转录程序的关键表观遗传因子。从机制上讲,HDAC介导的染色质结构改变能够激活由SRF和FOXM1诱导的促纤维原增生程序。HDAC还协调成纤维细胞促炎程序诱导LIF的表达,支持旁分泌促肿瘤的相互作用。在PDAC小鼠模型中,CAF中的HDAC缺失和HDAC抑制剂Ent处理可减少基质激活并抑制肿瘤进展。值得注意的是,HDAC抑制(HDACi)使脂肪生成成纤维细胞亚群富集,这是PDAC基质中肌成纤维细胞的潜在前体。总的来说,本项研究揭示了HDAC的基质靶向潜力,突出了这种表观遗传调节方法在PDAC治疗中的应用。
+ + + + +



