

 English
English文献解读|Nature(48.5):衰老通过激活整合应激反应促进转移
✦ +
+
论文ID
原名:Ageing promotes metastasis via activation of the integrated stress response
译名:衰老通过激活整合应激反应促进转移
期刊:Nature
影响因子:48.5
发表时间:2026.03.11
DOI号:10.1038/s41586-026-10216-0
背 景
衰老和癌症具有一些共同的核心特征,包括蛋白质稳态失衡、表观遗传重塑和营养感知改变,流行病学数据也表明,年龄增长与癌症发病率和死亡率升高相关。尽管存在这种关联,但衰老如何影响癌症进展的机制仍不甚明了。人类的年龄相关变化在小鼠中也基本保守,这使得小鼠成为研究哺乳动物衰老的理想模型。此外,基因工程小鼠模型(GEMM),例如KrasLSL-G12D/+; Trpflox/flox (KP)模型,能够在完整的微环境中忠实地重现人类肺肿瘤的发生发展,从而为研究肿瘤进展和扩散提供了一个生理相关的系统。越来越多的证据表明,肿瘤微环境的年龄相关改变可以驱动转移进展,但生理性衰老如何从内在层面重编程肺癌细胞以影响疾病进程仍不清楚。
实验设计
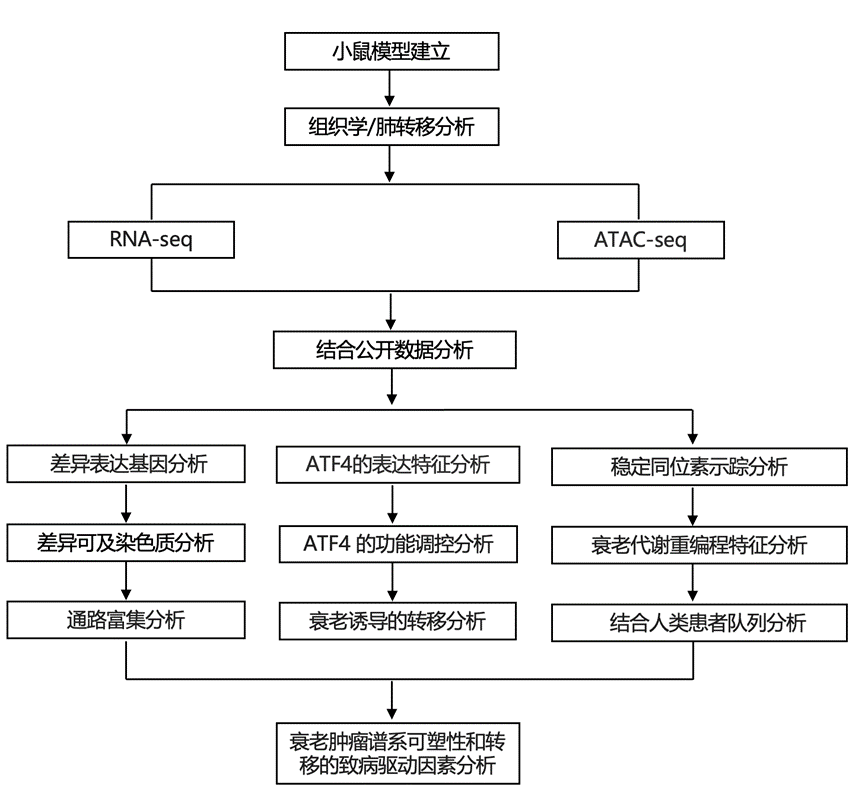
结 果
01

衰老会改变肺癌的进展
为了研究衰老对肺肿瘤发生的影响,研究团队通过气管内滴注编码 Cre 重组酶的病毒颗粒,同时在 2-3 月龄(KP-Young)和 18-19 月龄(KP -Old)的KP小鼠中诱导肺肿瘤(图1a)。这些年龄组分别代表了肺癌研究中常用的早期成年期和分子衰老表型开始显现的老年期。经过5-6个月的实验,老年组的年龄与非小细胞肺癌 (NSCLC) 的人类中位诊断年龄 70 岁相对应,从而准确地模拟了人类 NSCLC 最常见的患者群体特征,并克服了传统GEMM忽略宿主年龄的局限性。与KP-Young小鼠相比,KP-Old小鼠的原发性肺肿瘤负荷降低了2.5倍(图1b)。组织学和免疫组织化学(IHC)分析显示,KP-Old小鼠的肿瘤数量更少、体积更小、增殖能力更低。此外,KP-Old和KP -Young小鼠的所有肿瘤均表达肺泡2型(AT2)谱系标志物pro-SPC。与KP-Young小鼠相比,他们观察到KP-Old小鼠中遵循经典去分化轨迹的腺癌比例更高(图1c),表明虽然KP -Old小鼠的肺肿瘤负荷较低,但其恶性肿瘤进展却更快。使用 AT2 特异性 Adeno-SPC-Cre 证实,与KP -Young小鼠相比,KP-Old小鼠的肿瘤负荷降低,该 Adeno-SPC-Cre 仅在 AT2 细胞中重组KP。
纵向分析显示,KP -Young小鼠(140-357天)原发性肺肿瘤负荷随时间呈线性增加,而KP -Old小鼠在分析的时间点(106-247天)内肿瘤负荷保持较低水平,但转移发生较早且生存期缩短(图1d)。事实上,尸检结果显示,KP- Old小鼠局部淋巴结和远处器官的转移发生率显著高于KP -Young小鼠(图1e)。虽然在老年小鼠中未观察到雌雄小鼠肿瘤负荷和生存期的差异,但年轻雄性小鼠的生存期显著短于年轻雌性小鼠。
为了探究为何来自KP -Old 小鼠的较小且生长较慢的原发性肺肿瘤会更快地进展为转移性肺腺癌,他们接下来建立了来自KP -Young 和KP -Old 小鼠的原发性肿瘤培养物(分别记为KP-Y 和KP-O 培养物)。KP-Y 和KP-O 培养物的增殖速率相当,且 pro-SPC 水平相似,这与 AT2 来源相符。然而,KP-O 培养物中上皮-间质转化 (EMT) 标志物的表达增加(图1f),这与更强的转移特性相一致。接下来,他们研究了转移扩散的重要特征,包括在三维 (3D) 条件下的生长以及对失巢凋亡的抵抗力。KP-O培养物显示出更高的细胞活力(图1g)、更低的caspase 3/7活性以及在8天内更强的生长能力。为了模拟细胞外基质与肿瘤细胞的相互作用,将KP-O和KP-Y培养物包埋于Matrigel或胶原蛋白中。与KP-Y培养物相比,KP -O培养物在Matrigel中形成更大的球状体(图1h),在胶原蛋白中形成更具侵袭性的结构(图1i)。综上所述,这些数据表明,与KP -Y培养物相比,KP - O培养物在体外具有更高的转移倾向。
与KP-Y培养物相比,尾静脉注射KP -O培养物导致肺转移负荷增加,肾脏和心脏转移的发生率也更高(图1j-k)。这些结果强调了KP-O培养物的多样化趋向性,但并未进一步揭示其扩散和定植远处部位的能力。因此,他们将KP-O和KP-Y培养物皮下移植到小鼠侧腹,并定量分析了远处转移情况。组织学分析显示,KP-O培养物中远处肺转移灶更多,肺转移负荷也更高(图1l),这与其更强的转移能力相符。为了进一步评估其从起源组织播散、生长和扩散的能力,他们将KP-Y和KP-O培养物原位移植到小鼠肺部。KP -O培养物显示出显著更高的移植效率,更强的远处器官(包括肝脏、肾脏和脑)定植能力(80%的小鼠移植成功,而KP -Y组小鼠则无一例移植成功),以及受体小鼠生存时间的显著缩短(图1m-o)。与KP -Old小鼠的原发肿瘤一致,KP-O培养物的转移灶也表现出NKX2-1的缺失和HMGA2的获得(图1p)。综上所述,衰老会限制原发性肺肿瘤的生长,但有利于促进转移性肺腺癌的恶性进展。
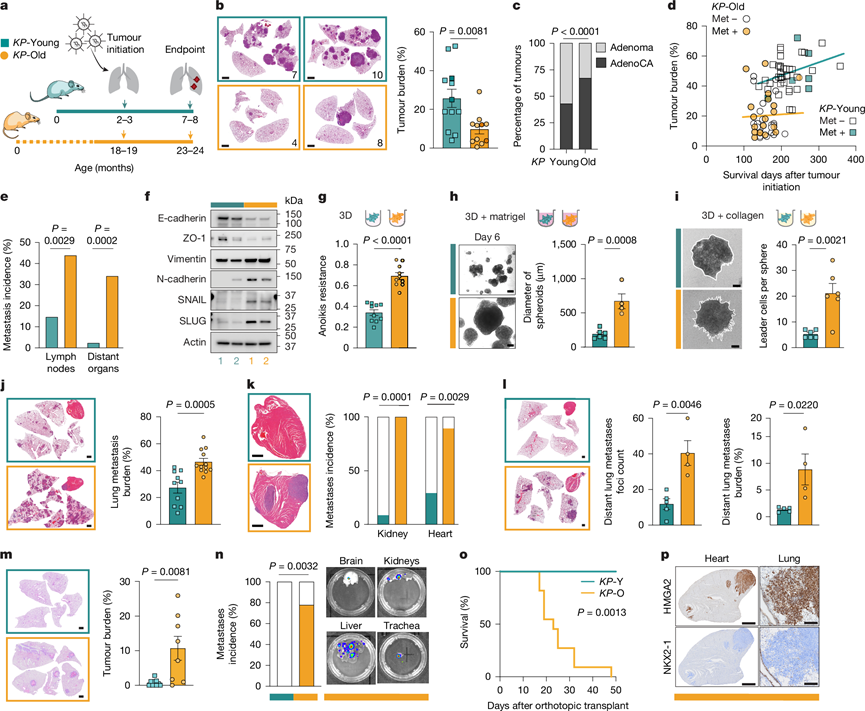
图1. 衰老有利于转移而不是原发性肺肿瘤生长。
(a) 实验时间线。(b) 代表性的苏木精-伊红 (H&E) 染色肺组织切片及肿瘤发生后 21 周的肿瘤负荷。(c) KP -Young 和KP -Old 肿瘤中腺瘤和腺癌 (AdenoCA) 的比例。(d-e) 局部淋巴结和远处转移的发生率。(f) Western blot分析。(g) 抗失巢凋亡能力。(h) 代表性球状体图像及球状体直径的定量分析。(i) 每个球状体中前导细胞的代表性图像及定量分析。(j) 肺组织切片经H&E染色显示肺转移负荷。(k) 心脏组织切片经H&E染色显示肾脏和心脏转移发生率。(l) 远处肺转移情况。(m) 肺组织切片经H&E染色显示肿瘤负荷。(n) 原位移植后的远处转移。(o) 原位移植小鼠的存活率。(p) KP-O形成的心脏(左)和肺(右)转移灶的代表性HMGA2和NKX2-1免疫组化染色。
02
衰老会增强 EMT 和 UPR 通路
与年龄相关的细胞变化通常通过染色质可及性的表观遗传改变进行调控。为了阐明老年小鼠肺肿瘤转移倾向增加的机制,他们对KP-Y和KP-O培养物进行了转录组分析(RNA-seq)和转座酶可及染色质测序(ATAC-seq),结果在KP-O培养物与KP-Y培养物相比,鉴定出2022个差异表达基因(DEG)和27206个差异可及染色质区域。通路富集分析[基因集富集分析(GSEA)、RNA-seq和基因组区域富集注释工具(GREAT)、ATAC-seq]鉴定出与转录和表观遗传改变相关的关键通路。在前八个富集通路中,出现了两个主要且重叠的主题:EMT 和未折叠蛋白反应 (UPR)(图2a),两者在KP -O 培养物中均高度富集(图2b,图S4a-d)。
UPR通过三个分支传递内质网应激信号:IRE1α(由ERN1编码)–XBP1、ATF6和PERK–eIF2α–ATF4。对KP -O培养物中富集的DEG进行STRING分析发现,包含ATF4的网络显著富集,而IRE1α和ATF6缺乏任何功能关联,这强烈提示KP-O培养物中UPR–PERK–ATF4通路激活。UPR通过PERK介导的eIF2α磷酸化参与整合应激反应(ISR),最终诱导ATF4的表达,ATF4是ISR的主要转录效应因子(图2c)。
为了探究衰老如何调控这些通路,将原代培养细胞暴露于低葡萄糖条件下6-24小时。低葡萄糖条件是扩散性肿瘤细胞所面临的生理相关营养胁迫,已知该胁迫也会诱导内质网应激。在低葡萄糖条件下,与KP-Y培养物相比,KP -O培养物中UPR-ISR的PERK分支激活显著增强,PERK(p-PERK)、eIF2α(p-eIF2α)的磷酸化水平升高,ATF4蛋白水平也升高(图2c,d)。值得注意的是,在KP-O培养物中,葡萄糖剥夺后p-eIF2α水平仍然升高,与ATF4的诱导同时发生。然而,由于GADD34的反馈调节,即使p-eIF2α水平下降,ATF4蛋白的表达仍然持续存在(图2c-d)。即使是轻微的环境波动,例如在标准高葡萄糖条件下更换培养基,也会引发 p-eIF2α 的增加(图2d),这表明KP-O 培养物中 ISR 激活的阈值较低。
在正常培养条件下接种48小时后,主要上游ISR激酶(GCN2、PKR、PERK、HRI)和ISR核心蛋白eIF2α的磷酸化水平相似(图2c),但ATF4蛋白及其下游靶标(包括p-4EBP1、ASNS、SLC7A11、CHOP和BCAT1)在KP-O培养物中仍显著高于KP -Y培养物。KP-O和KP-Y培养物中ATF4蛋白的稳定性相当。虽然在转录抑制后,KP-O培养物中Atf4 mRNA的衰减速度更快,但转录抑制后,KP-Y和KP -O培养物中的ATF4蛋白均迅速耗竭(图2f)。相比之下,蛋白酶体抑制导致KP -O培养物中ATF4积累量更高(图2f),这与转录和/或翻译增强相一致,但当转录阻断时,ATF4蛋白水平却无法恢复。这些数据表明,KP -O培养物中ATF4水平升高主要反映的是转录增强,而非mRNA稳定性或蛋白质周转的改变。总而言之,这些发现表明,通过允许染色质状态增强的转录和增强的ISR信号共同驱动了KP -O培养物中观察到的持续升高的ATF4水平。
ATF4与细胞凋亡抵抗和转移有关,其表达与人类癌细胞系的肝转移渗透率密切相关。与此一致的是,KP-Old小鼠肿瘤中ATF4阳性细胞核的百分比显著高于KP -Young小鼠(图2g)。
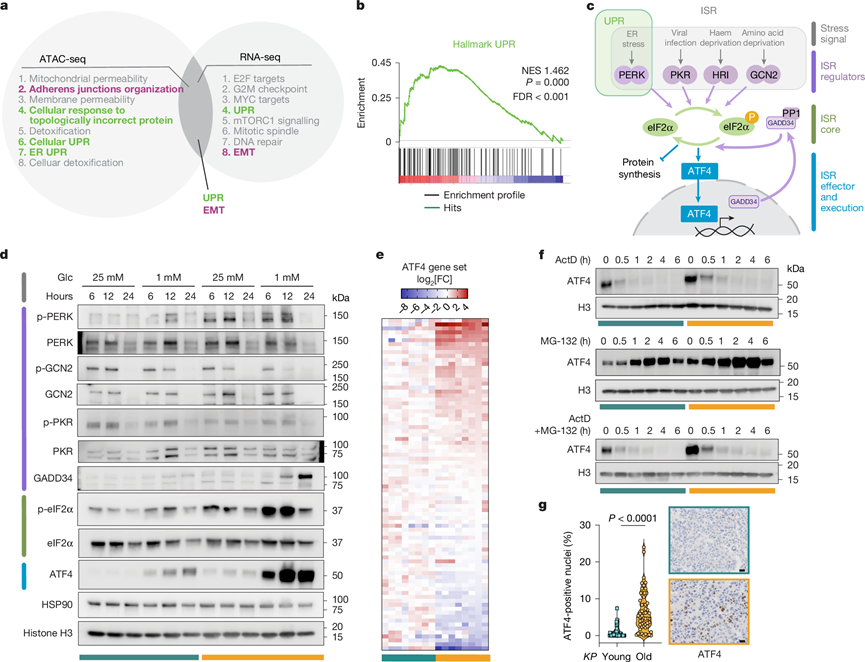
图2. 肿瘤中 UPR-ISR-ATF4 的持续表观遗传诱导。
(a) 通过对ATAC-seq进行GREAT分析以及对KP-O和KP-Y原代培养物进行RNA-seq进行GSEA分析鉴定出的主要富集通路。(b) GSEA富集图。(c) ISR通路示意图。(d) 在培养基更换后指定时间点,分别在高糖和低糖培养基中培养KP-Y和KP-O的Western blot结果。(e) KP -Y和KP -O中ATF4靶基因表达的热图。(f) Western blot分析。(g) 肺肿瘤中 ATF4 阳性细胞核的定量分析。
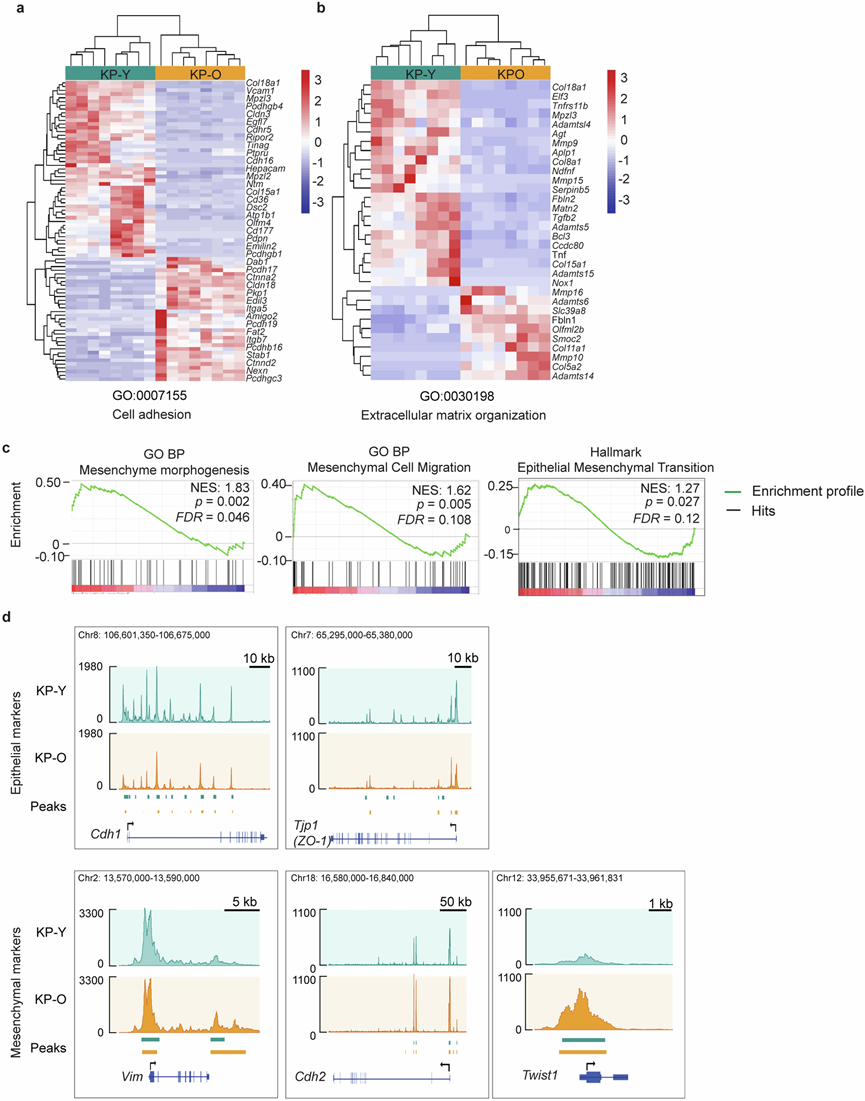
图S4. 转移相关通路在KP -O 原代培养物中富集。
(a-b) 基因热图分析。(c) 间充质形态发生、间充质细胞迁移和上皮间质转化的 GSEA 富集图。(d) 染色质可及性分析。
03
ATF4 驱动衰老诱导的转移
ATF4与细胞凋亡抵抗和转移有关,其表达与人类癌细胞系的肝转移渗透率密切相关。与此一致的是,KP-Old小鼠肿瘤中ATF4阳性细胞核的百分比显著高于KP -Young小鼠(图2g)。为了确定ATF4是否在衰老诱导的转移中起因果作用,他们首先使用整合应激反应抑制剂(ISRIB)(一种ISR抑制剂)对ATF4翻译进行药理学抑制,该抑制剂可降低KP-O培养物中ATF4蛋白水平及其下游靶基因的表达(图3a)。长期使用 ISRIB 处理,在不影响贴壁细胞活力的情况下,降低了KP-O 培养物的抗失巢凋亡能力、克隆形成能力和体内转移倾向(图3b-c)。类似地,PERK 抑制剂也降低了抗失巢凋亡能力,表明 PERK-ATF4 轴在这种衰老诱导的表型中发挥作用。接下来,他们通过 CRISPR-Cas9 介导的基因敲除(靶向Atf4 的单向导 RNA)和强力霉素诱导的基于mirE 的短发夹 RNA (shRNA) 介导的 ATF4 耗竭(图3d)对 ATF4 进行基因靶向。在KP-Y和KP -O两种培养物中,ATF4及其下游靶基因(包括ASNS、SLC7A11和4EBP1)的蛋白水平均降低(图3d)。值得注意的是,在人肺腺癌(LUAD)细胞系中,ASNS和SLC7A11的表达与ATF4的表达呈显著正相关,而4EBP1的表达则无此相关性,提示前两者可能参与ATF4驱动的转移。与此相符的是,KP-Old小鼠的肿瘤组织中ASNS和SLC7A11均呈强阳性染色。此外,敲除或敲低Atf4降低了KP-O培养物而非KP-Y培养物的抗凋亡能力和适应性,并显著下调了EMT标志物。为了验证KP-O小鼠肿瘤细胞转移能力的增强是否在体内依赖于ATF4,他们分析了KP-O和KP-Y小鼠的肿瘤细胞。将ATF4缺失或敲除的KP-O细胞培养物通过尾静脉注射进行移植。与未靶向的KP -O对照组相比,KP-O细胞培养物中Atf4的缺失或敲低显著降低了肺转移负荷(图3e-f和)。相反,无论是否靶向ATF4,注射KP -Y细胞培养物的小鼠与注射KP -O细胞培养物的小鼠相比,肺转移负荷极低(图3e-f)。总之,这些结果表明ATF4调控原代KP -O细胞培养物中衰老诱导的转移潜能。
为了评估 ATF4 是否足以驱动转移,他们在KP-Y 和KP-O 细胞培养物中过表达了 ATF4(图3g)。ATF4 过表达导致KP-Y 和KP-O 细胞中 ATF4 水平出现类似的轻微升高,同时 ATF4 靶基因(ASNS、SLC7A11、CHOP 和 BCAT1)也上调。值得注意的是,在KP-Y细胞培养物中过表达ATF4也诱导了 EMT 标志蛋白的表达(图3g)。KP-Y细胞培养物中ATF4过表达导致1661个基因的差异表达,其中过表达ATF4的KP-Y 和KP-O细胞与KP -Y细胞转录组之间存在 592个差异表达基因的重叠。GSEA分析发现,ATF4过表达组中UPR和EMT通路富集(图3h),与KP-O组中观察到的富集情况相似,进一步证实了ATF4在衰老诱导的转移中的作用。功能上,KP-Y组中ATF4过表达使细胞抗凋亡能力和转移能力均达到与KP -O组相似的水平(图3i)。在人A549肺腺癌细胞中,ATF4过表达足以显著增加肺转移负荷,并促进癌细胞在肺外部位(如心脏)的定植(图3j-l)。因此,ATF4是衰老诱导转移状态的必要且充分条件。
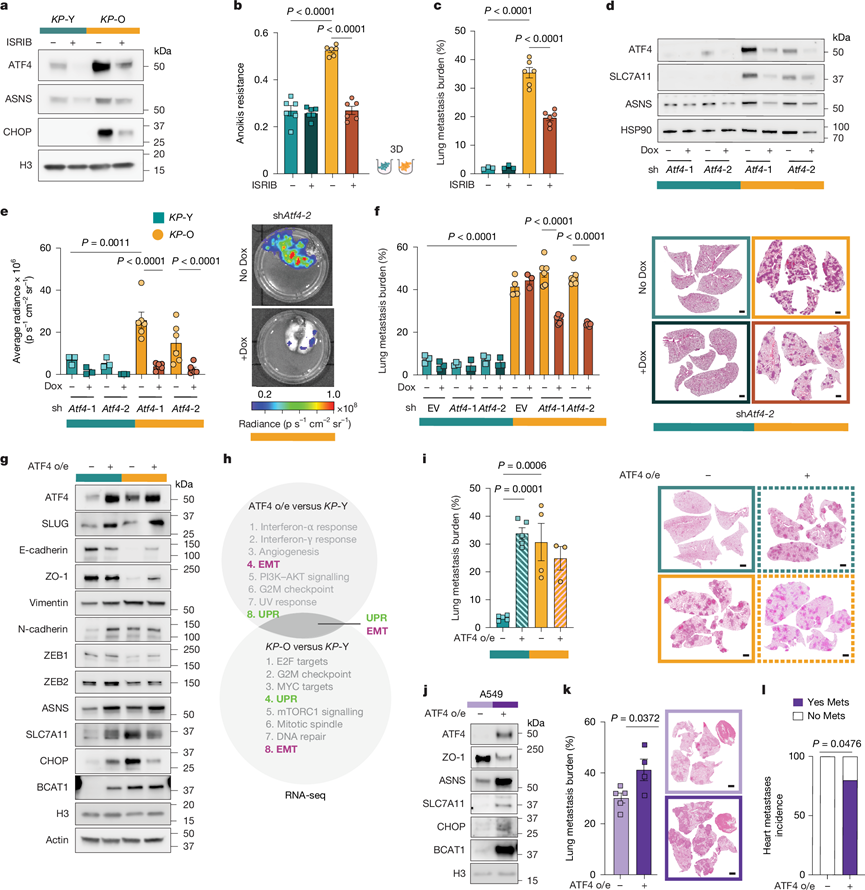
图3. ATF4 诱导的可塑性是驱动转移的必要和充分条件。
(a) Western blot结果。(b) 抗失巢凋亡能力分析。(c) 肺转移负荷。(d) Western blot结果。(e-f) 肺转移负荷。(g) Western blot分析。(h) GSEA分析中富集通路最多的通路。(i) 静脉注射表达对照或ATF4过表达/表达(o/e)的KP-Y和KP-O细胞后,肺转移负荷及代表性H&E染色肺组织切片。(j) A549对照细胞和ATF4过表达/表达细胞中ATF4、ATF4靶基因和EMT标志物的Western blot分析。(k) 肺转移负荷及代表性H&E染色肺组织切片。(l) 注射A549对照细胞或ATF4过表达/表达细胞的小鼠心脏转移发生率。
04
ATF4驱动的衰老代谢重编程
为了研究代谢应激反应和细胞代谢的关键调控因子ATF4是否会改变KP-O培养物的代谢,他们使用[1,2-13C] -D-葡萄糖进行了稳定同位素示踪。KP-O培养物显示出糖酵解通量增加,这体现在通过糖酵解而非磷酸戊糖途径产生的丙酮酸和乳酸的标记水平升高。磷酸戊糖途径通过维持氧化还原稳态来支持转移性癌细胞(图4a-c)。相反,KP -O培养物中葡萄糖来源的补充物质进入三羧酸循环(TCA循环)的量减少。葡萄糖补充途径通过丙酮酸脱氢酶途径进入三羧酸循环,支持能量产生;或者通过丙酮酸羧化酶途径补充生物合成中间体。使用[3-13C]-葡萄糖进行示踪显示,KP-O培养物中丙酮酸羧化酶介导的碳掺入显著降低,表明代谢途径发生了转变,不再依赖葡萄糖补充途径。因此,他们推测KP -O细胞可能表现出代谢可塑性,更多地依赖其他代谢途径来维持三羧酸循环中间体。与此相符的是,使用[U13C]-L-谷氨酰胺进行稳定同位素示踪显示,与KP-Y培养物相比,KP -O培养物中谷氨酰胺衍生的补充途径增加(图4d-e)。这种代谢转变还得到了KP -O与KP-Y相比细胞外酸化速率显著增加的进一步支持,而耗氧速率没有变化,这与有氧糖酵解和谷氨酰胺补充代谢的增加相一致(图4f-g)。在Atf4缺陷的KP -O培养物中,谷氨酰胺对TCA中间体的贡献显著降低(图4h),而在KP -Y培养物中过表达ATF4后,谷氨酰胺对TCA中间体的贡献显著增加(图4i),表明谷氨酰胺分解的增强是KP-O培养物中ATF4依赖性的特征。
接下来,他们评估了KP -O培养物中ATF4驱动的代谢可塑性是否可用于治疗。初步药物筛选显示,KP-O培养物对SLC7A11、BCAT或PHGDH抑制剂的敏感性无差异。然而,KP-O培养物对谷氨酰胺耗竭和谷氨酰胺类似物DON更为敏感,但对糖酵解抑制剂2-脱氧葡萄糖(2-DG)则不敏感。因此,他们检测了能够阻断谷氨酰胺转化为谷氨酸(即谷氨酰胺分解)这一限速步骤的GLSi。KP-O 培养物对两种独立的 GLSi,即 CB-839(特拉格列那司他)和 BPTES,表现出明显的敏感性(图4j),这与之前高 SLC7A11 水平与 GLS 抑制敏感性之间的联系一致。此外,KP-O 培养物比KP-Y 培养物对 V-9302(一种主要谷氨酰胺转运蛋白 ASCT2 的拮抗剂)更敏感。
为了确定KP -O培养物对GLSi敏感性的来源,他们用多种代谢物、氨基酸、抗氧化剂和小分子预处理了KP-O和KP -Y培养物。细胞可渗透的α-酮戊二酸(α-KG)类似物二甲基-2-氧代戊二酸(DMG)和葡萄糖衍生的三羧酸循环碳源丙酮酸均能挽救KP-O培养物对CB-839的敏感性(图4k)。除半胱氨酸-谷氨酸逆向转运系统抑制剂erastin外,其他所有测试的代谢物或抑制剂均不能挽救KP -O培养物对CB-839的敏感性,这强烈表明KP -O培养物对谷氨酸水平降低高度敏感(图4k)。接下来,他们检测了ATF4是否介导KP -O对GLSi的敏感性。事实上,用低剂量ISRIB预处理或基因敲除ATF4的KP -O培养物丧失了对CB-839的敏感性。相应地,ATF4过表达使先前具有抗性的KP -Y培养物对CB-839敏感。在3D培养中,用GLSi或谷氨酰胺转运体抑制剂V-9302处理后,KP -O球体丧失了抗失巢凋亡能力(图4l-m)。值得注意的是,球体对CB-839的敏感性依赖于ATF4,因为短期ISRIB处理本身不足以改变其抗失巢凋亡能力,但却能挽救其存活。此外,ATF4 过表达赋予KP -Y 球体抗凋亡能力,而新获得的 CB-839 敏感性则逆转了这种能力(图4l)。
此外, DMG、丙酮酸和erastin可以挽救KP -O细胞中CB-839诱导的抗凋亡能力丧失,但其他干预措施则无效(图4m)。与这些结果一致,ATF4表达与转移性人肺腺癌(LUAD)细胞系(而非原发性LUAD细胞系)的CB-839敏感性相关。
为了验证他们研究结果的体内相关性,他们将KP-O和KP -Y细胞培养物分别通过静脉注射或皮下注射移植到小鼠体内,并随机分为CB-839组和载体组。静脉注射后,载体组小鼠体内KP -O细胞培养物形成了大量肺转移灶,而CB-839组小鼠体内几乎检测不到转移灶(图4n-o)。相比之下,无论是否接受治疗,注射KP -Y细胞的小鼠几乎没有发生转移(图4n-o)。皮下移植后,CB-839治疗并未影响KP -O和KP -Y细胞的肿瘤生长速度(图4p)。虽然KP-O肿瘤总体上显著大于KP -Y肿瘤,但CB-839治疗并未改变各组最终肿瘤的重量。CB-839治疗几乎完全抑制了KP-O肿瘤的远处转移,这通过远处转移灶计数和肺转移负荷评估得出,且不影响KP -O肿瘤的生长或最终大小(图4p-q)。相比之下,KP-Y肿瘤及其转移不受GLSi的影响(图4p-q)。总而言之,衰老诱导的ATF4介导的代谢重编程构成了转移性疾病中一个可药物靶向的脆弱点,可作为老年NSCLC患者的潜在辅助治疗靶点。
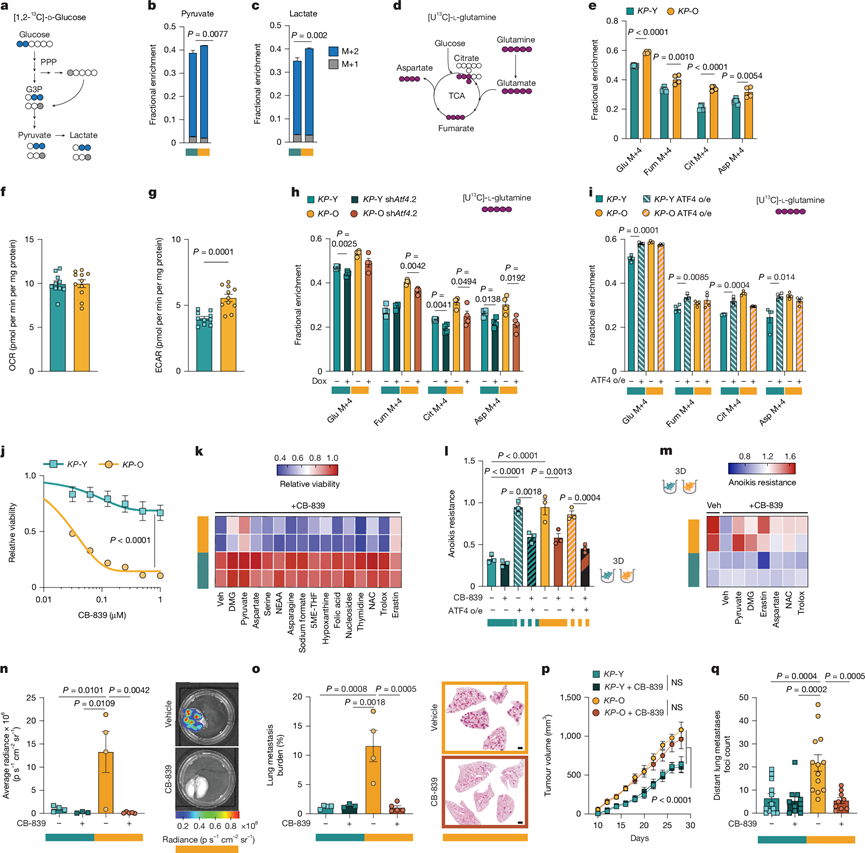
图4. 靶向 ATF4 诱导的代谢可塑性可阻断转移。
(a) 示踪示意图。(b-c) 标记的丙酮酸和乳酸的分数。(d) 谷氨酰胺示踪示意图。(e) 同位素异构体质量分析。(f-g) 耗氧率(OCR)和细胞外酸化率(ECAR)。(h-i) 质谱同位素 分析。(j) 经 CB-839 处理的KP -Y 和KP -O的相对存活率。(k) 经 0.1 μM CB-839 和所示化合物处理的KP -Y 和KP -O的相对存活率热图。(l-m) 抗失巢凋亡能力。(n-o) 肺转移负荷。(p-q) 皮下肿瘤的纵向生长和肺转移灶的 H&E 定量。
05
衰老会改变人类非小细胞肺癌的进展
为了评估从KP- Old小鼠中获得的研究结果与NSCLC患者临床实际情况的相关性,他们从2016年至2018年间瑞典西部西约塔兰省和哈兰省所有确诊NSCLC患者的病历中提取了年龄、诊断分期和KRAS突变状态的数据(图5a)。其中,368例(36.9%)患者携带KRAS突变(KRAS MUT),而629例患者未携带KRAS突变(KRAS WT)。整个队列的中位诊断年龄为71岁,两个突变组的中位诊断年龄也相同(图5b)。接下来,他们将该队列分为两个年龄组进行分析:60岁以下患者组和65-75岁老年患者组。后一组的年龄与安乐死时KP-Old小鼠的年龄(±5岁)相符,并且与该队列的中位诊断年龄以及所有KRAS突变患者中的最大比例(65.2%)一致。观察到65-75岁组的KRAS突变发生率高于年轻组(图5c),这一发现也见于MSK-IMPACT 数据集(n =435)。此外,与年轻(60岁以下)的KRAS突变患者相比,65-75岁的KRAS突变患者更易出现局部区域进展和转移性疾病(IIIb-IV期NSCLC)。在KRAS野生型患者中未观察到这种模式(图5d)。根据致癌驱动基因状态(EGFR突变、其他已知的驱动基因和未知驱动基因)将KRAS野生型组进一步细分为亚组,结果显示,未知驱动基因患者的年龄依赖性模式与KRAS突变组相似。相比之下,携带EGFR突变或其他已知驱动基因改变的患者则呈现出不同的分布,这表明观察到的与年龄相关的疾病进展不仅限于KRAS突变,可能还特异于某些遗传亚组。
为了探究肿瘤特征与年龄的关系,他们接下来从至少记录了两次计算机断层扫描测量结果的患者病历中提取了原发肿瘤的测量数据(n = 464)(图5a)。其中,172 例患者携带KRAS突变(KRAS MUT),292 例患者未携带 KRAS 突变(KRASWT)。在KRASMUT NSCLC伴局部区域晚期和转移性疾病(IIIb-IV 期)的患者中,随着诊断年龄的增长,原发肿瘤的大小明显减小(图5e),这与易转移KP-Old 小鼠的情况相似(图1c)。未在早期KRAS MUT或KRASWT NSCLC以及所有分期中驱动基因突变未知的NSCLC患者中观察到类似的趋势。然而,在ALK阳性NSCLC患者中观察到了类似的肿瘤大小随年龄增长而减小的现象。综上所述,这些发现表明,虽然KP -Old小鼠原发性肺肿瘤负荷降低,但转移发生率却增加,这反映了65-75岁KRASMUTNSCLC患者的临床实际情况,并且可能也适用于其他致癌驱动基因的患者。支持这一结论的是,来自许多公开在线数据存储库的数据表明,在一组人类肺癌细胞系中,倍增时间(小时)与年龄(年)之间存在正相关性,同时 EMT 标志物上调,这与在KP -O 培养物中的发现一致(图1e-f)。
ATF4是衰老诱导转移的关键驱动因素,其表达与老年KP -Old小鼠的侵袭性疾病相关。鉴于在65-75岁KRAS突变患者中观察到转移发生率增加,他们提出ATF4可能作为晚期疾病进展的生物标志物。为了验证这一假设,他们分析了癌症基因组图谱-肺腺癌队列(TCGA-LUAD)数据集,并根据肿瘤分期和ATF4表达对患者进行分层。虽然在早期(I-II期)患者中未观察到显著的生存差异(图5f),但在晚期(III-IV期)患者中,高ATF4表达与生存期缩短相关(中位生存期分别为1.28年和2.61年)(图5g),这进一步证实了ATF4在人类转移性疾病中的重要性。最后,为了直接评估ATF4的临床意义,他们检测了来自瑞典队列37例KRAS突变型肺腺癌(LUAD)切除患者的肿瘤组织中ATF4的表达。在该队列中,ATF4核阳性与诊断年龄呈显著正相关(图5h)。按年龄分层分析证实,65岁以上患者的ATF4水平显著高于60岁以下患者(图5i)。此外,高ATF4水平与总生存期显著降低相关,高ATF4水平患者的5年生存率仅为32.6%,而低ATF4水平患者的5年生存率为65.7%。8年时,生存率差异更为显著(图5j)。少数ATF4高表达肿瘤的年轻患者生存期也较短。这些数据表明,ATF4是KRAS突变型肺腺癌患者预后不良的可靠标志物。总而言之,这些发现凸显了ATF4作为治疗靶点在缓解NSCLC中由衰老驱动的转移进展方面的潜力。由于老年人占肺腺癌患者的大多数,因此,ATF4的年龄相关性激活成为转移的驱动因素,也是最具代表性的患者群体中一个可进行治疗的脆弱点。
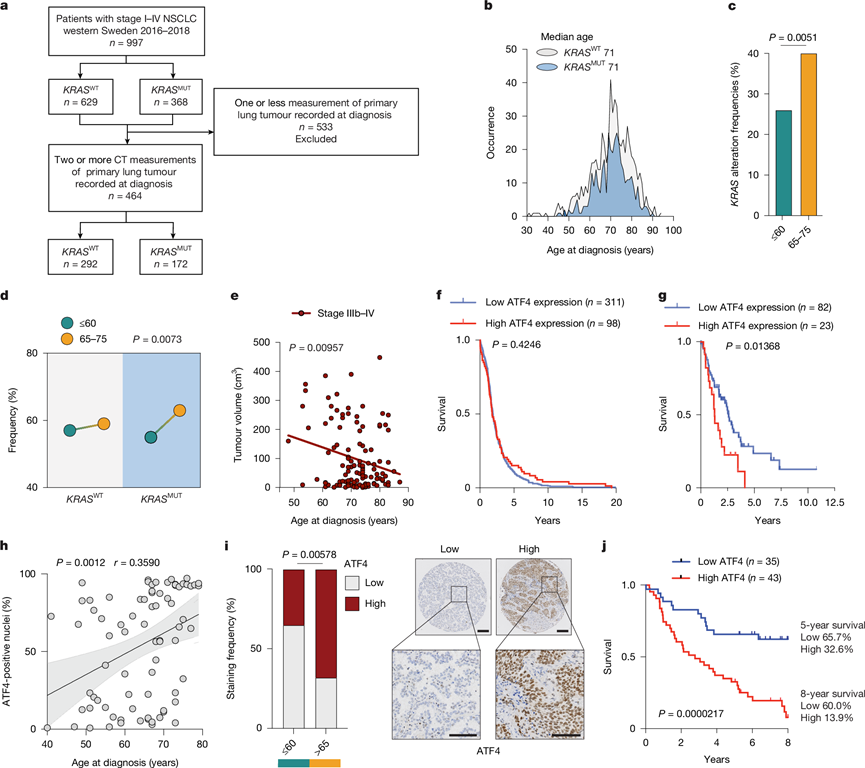
图5. 人类非小细胞肺癌重现了衰老表型。
(a) 研究患者选择流程图。(b) 患者的诊断年龄分布。(c) 瑞典西部队列中年轻(60 岁或以下)和年长(65-75)患者诊断时 KRAS 突变频率的发生率。(d) 携带 KRAS 突变(KRASMUT)或未携带 KRAS 突变(KRASWT)的比例。 (e) 相关性分析。(f-g) 肺腺癌患者的总生存期。(h) 来自瑞典 NSCLC-TMA 队列的KRAS突变型肺腺癌患者中 ATF4 阳性细胞核百分比与诊断年龄之间的相关性。(i) 患者肿瘤中ATF4染色强度。(j) 腺癌患者的生存情况。
+ + + + + + + + + + +
结 论
本研究表明,衰老会重编程KRAS驱动的肺腺癌的演化轨迹,通过表观遗传激活整合应激反应(ISR)来限制原发肿瘤的生长,同时促进转移扩散。ISR效应因子ATF4驱动上皮和代谢可塑性,赋予肿瘤转移能力。机制上,衰老的肿瘤细胞对未折叠蛋白反应的PERK-eIF2α通路表现出更高的敏感性,从而维持持续的ATF4信号传导。通过基因或药物靶向ISR-ATF4可以消除这些适应性改变并限制肿瘤扩散,而ATF4的过表达本身就足以诱导转移。衰老-ATF4轴对谷氨酰胺代谢的依赖性揭示了一个具有治疗意义的脆弱性。临床分析证实,ATF4在老年肿瘤中富集,且与生存率低和疾病分期晚相关。综上所述,这些结果表明,表观遗传ISR-ATF4激活是老年肿瘤谱系可塑性和转移的致病驱动因素,为老年肺腺癌患者(肺癌中最常见但研究不足的亚型)的治疗提供了契机。
+ + + + +



