

 English
English文献解读|Genome Biol(10.1):时空建模揭示胶质母细胞瘤的高分辨率侵袭状态
✦ +
+
论文ID
原名:Spatiotemporal modeling reveals high-resolution invasion states in glioblastoma
译名:时空建模揭示胶质母细胞瘤的高分辨率侵袭状态
期刊:Genome Biology
影响因子:10.1
发表时间:2024.10.10
DOI号:10.1186/s13059-024-03407-3
背 景
胶质母细胞瘤(GBM)是成人中最常见的恶性脑肿瘤,虽然采用了多模式治疗,包括最大程度的安全手术切除、放疗和化疗,但仍无法治愈。胶质母细胞瘤的治疗困难部分源于其高度侵袭性的表型,其中单个肿瘤细胞弥漫性地在正常组织中移动或沿着血管周围路径或白质束扩散到远离主要肿瘤块的地方。虽然手术切除了大部分肿瘤,但这些浸润细胞仍有保留,因为它们不仅难以检测,而且如果不产生不可接受的神经系统后果就无法去除。残留的细胞可以继续进化并适应传统和合理疗法的选择压力——这个过程是多方面的,涉及遗传异质性、表型可塑性以及与肿瘤微环境 (TME) 互动并将其纳入促肿瘤状态的能力——从而导致复发。因此,了解侵袭前沿的生物学特性并阐明这些细胞与周围正常细胞和环境相互作用以促进恶性表型的机制具有很高的临床意义。
实验设计
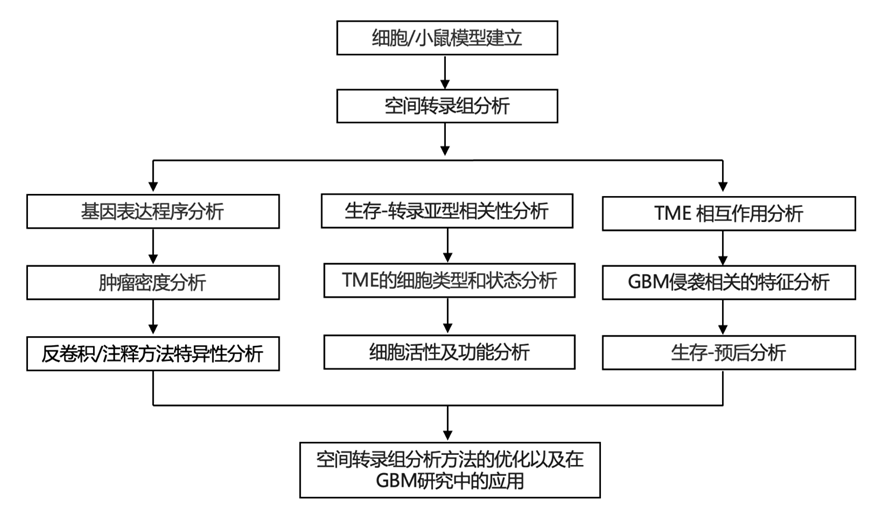
结 果
01

GBM 异种移植模型中的空间转录组分析
为了全面捕获体内空间转录异质性,研究团队从手术样本中选择了六种在机构建立的、特征明确的脑肿瘤起始细胞 (BTIC) 系。这些细胞来自四名患者,涵盖了一系列临床变量,包括性别、是否接受标准疗法、MGMT 甲基化状态和常见的 GBM 分子驱动因素(图1a-b)。除了遗传驱动因素的多样性之外,本研究的队列还包括来自两名患者(BT143 和 BT238)的 BTIC 系,这些系捕获了肿瘤演变过程中获得的表型多样性。在这些患者中,多条细胞系同时来自术前 MRI 定义的肿瘤解剖上不同的区域,包括密集生长的核心 (x)、对比增强的高度血管化的肿瘤边缘 (y) 和肿瘤高度弥散的前缘 (z),通常位于手术边缘之外。来自每位患者的细胞系共有基因组驱动因子,但在体内保持不同的生长表型,其中 x 细胞系相对于弥漫浸润的 y 和 z 细胞系生长密集(图1c)。对同样来自边缘与核心手术样本的其他细胞系以及在适应缺氧的背景下也进行了类似的观察。队列内和队列间 x/y/z 生长模式的一致性表明,GBM 细胞在肿瘤进化过程中可以获得可预测的表型适应,这些适应与空间环境(例如,核心与边缘)有关,并且它们是可遗传的,可能在基因组或表观基因组中固定。因此, x/y/z BTIC 模型为研究密集生长与弥漫生长背后的表达程序提供了机会。最后,为了捕捉不同肿瘤阶段的侵袭动态,该队列根据已知的终点时间(跨系范围在 76 至 428 天之间)包括早期、中期和晚期生长时间点(图1 b)。
他们利用 Visium 平台从总共 23 个样本(图 1 c)中收集了 51952 个单独的空间转录组 分析 (stRNA-seq) 数据(点)(图 1 c),涵盖所有细胞系和时间点。在许多情况下,他们能够将完整的冠状脑切片对角线地放入捕获区域内,从而确保跨整个肿瘤和侵入前沿进行分析。在其他情况下,注射侧 (i) 和对侧 (c) 安装在单独的捕获区域(例如,BT143y/z 端点样本)(图 1 c)。他们使用人与鼠转录组混合数据来分析肿瘤细胞对每个点转录输出的相对贡献。混合度范围从 100% 小鼠细胞点的 0 到 100% 肿瘤细胞点的 1(图 1c-e)。能够区分小鼠细胞和人类细胞的灵敏度使他们将析重点放在高(80-100%;D4)、中高(50-80%;D3)、中低(20-50%;D2)和低(5-20%;D1)肿瘤细胞密度区域,以及无肿瘤的小鼠脑点(D0)。他们观察到以这种方式定义的肿瘤区域中的线和时间点之间的肿瘤密度水平差异很大。BT143x 脱颖而出,成为生长最密集的线,许多斑点在终点时的肿瘤密度 > 80%(图 1 e)。BT161 和 BT238 是第二密集的线,许多斑点在终点时的肿瘤细胞密度为 50-80%。相比之下,BT134 终点肿瘤和所有早期时间点的跨线肿瘤均呈弥漫性生长。
由于 stRNA-seq 不具备单细胞分辨率,他们使用无监督反卷积分别识别人类和小鼠数据的混合细胞类型和状态。使用共识非负矩阵分解 (cNMF)在所有 23 个样本中识别出稳健的转录程序,并定量它们在点内的相对使用情况。分解产生了 15 个人类肿瘤细胞程序(称为h1-h15)和 90 个小鼠大脑和 TME 程序(称为m1-m90),这与小鼠大脑的转录多样性更高一致。为了阐明程序是否代表细胞类型、细胞状态或解剖结构,他们 (i) 使用已建立的正常小鼠脑细胞类型、脑结构、TME 特异性细胞类型和人类 GBM 状态的参考标记基因,(ii) 评估程序特定于生物途径的富集,以及 (iii) 将程序使用的空间模式与来自 Allen Brain Atlas 通用坐标框架 (CCFv3) 的已知小鼠大脑结构进行比较。
人类肿瘤程序在核心中最为多样化,高密度区域每个点有 3 到 4 个可辨别的程序,低密度区域每个点有 1 到 2 个程序。小鼠程序包括 11 种细胞活性和 18 种正常大脑中的细胞类型、19 种 TME 特异性或富集的细胞类型和状态,以及 42 种代表细胞类型组合的程序,这些程序无法在此分解级别进一步消除歧义。与 ABA 的比较分析证实,这些组合程序中的大多数对应于小鼠大脑的解剖区域或结构。他们在无 GBM 的区域(D0)和肿瘤前缘(D1)中检测到平均每个点 5 个正常大脑小鼠程序,在较高的肿瘤密度下多样性降低。相比之下,肿瘤区域每个点仅观察到 1 到 2 个 TME 相关程序。总体而言,在人类和小鼠数据中,他们的方法能够定量每个点最多 8 个程序的使用情况,灵敏度远高于之前使用 stRNA-seq 分析实现的灵敏度(即每个点约 2 个程序)。
为了确保本研究的工作流程能够产生有意义的结果,他们评估了一组预计在细胞类型和解剖结构方面与先前文献非常接近的小鼠程序。第一个例子是程序m25,根据标记基因的富集,它与室管膜细胞相对应。室管膜细胞形成局限于心室内膜的单细胞层,事实上,m25 定位于心室内膜,其他地方没有背景信号(图1 f-g)。使用值介于 0.3 和 0.5 之间,表明室管膜细胞占这些点信号的三分之一到二分之一,与心室内膜中已知的细胞组成一致。标记基因和转录因子(TF)活性的分析进一步验证了m25为室管膜程序(图1 h-i)。在脑室下区域,他们还鉴定了每个点的细胞频率较低(3-10%)的祖细胞程序,包括集中在背外侧脑室区域的胶质生成祖细胞(m58),以及两个位于侧脑室内膜并弥漫扩散到脑实质中的增殖祖细胞程序(m6、m83)(图1f-g)。他们可以区分周细胞和内皮细胞,即使这些细胞类型在血管内空间上总是同时出现,在给定的 Visium 点内仅占少数信号(周细胞程序的最大使用率为 8%),并且以前没有在空间数据集中反卷积。他们重点分析了皮质层(以高分辨率识别),包括最外层的脑膜(m4)和血管软脑膜平滑肌细胞(m11)。皮质层以空间重叠的方式排列,反映了沿大脑径向轴的细胞类型和状态的变化组成。该组成梯度由程序使用值定量捕获,重现了皮质程序之间的空间重叠水平。基于这些观察,本研究的反卷积和注释方法具有高度特异性(能够识别独特的细胞类型、状态和解剖结构),高度灵敏(能够对整体信号较低的信号进行反卷积在 3-10% 的范围内),可以识别空间相干和弥散程序,并提供可解释的使用值,可定量每个 Visium 点内的最多八个程序。
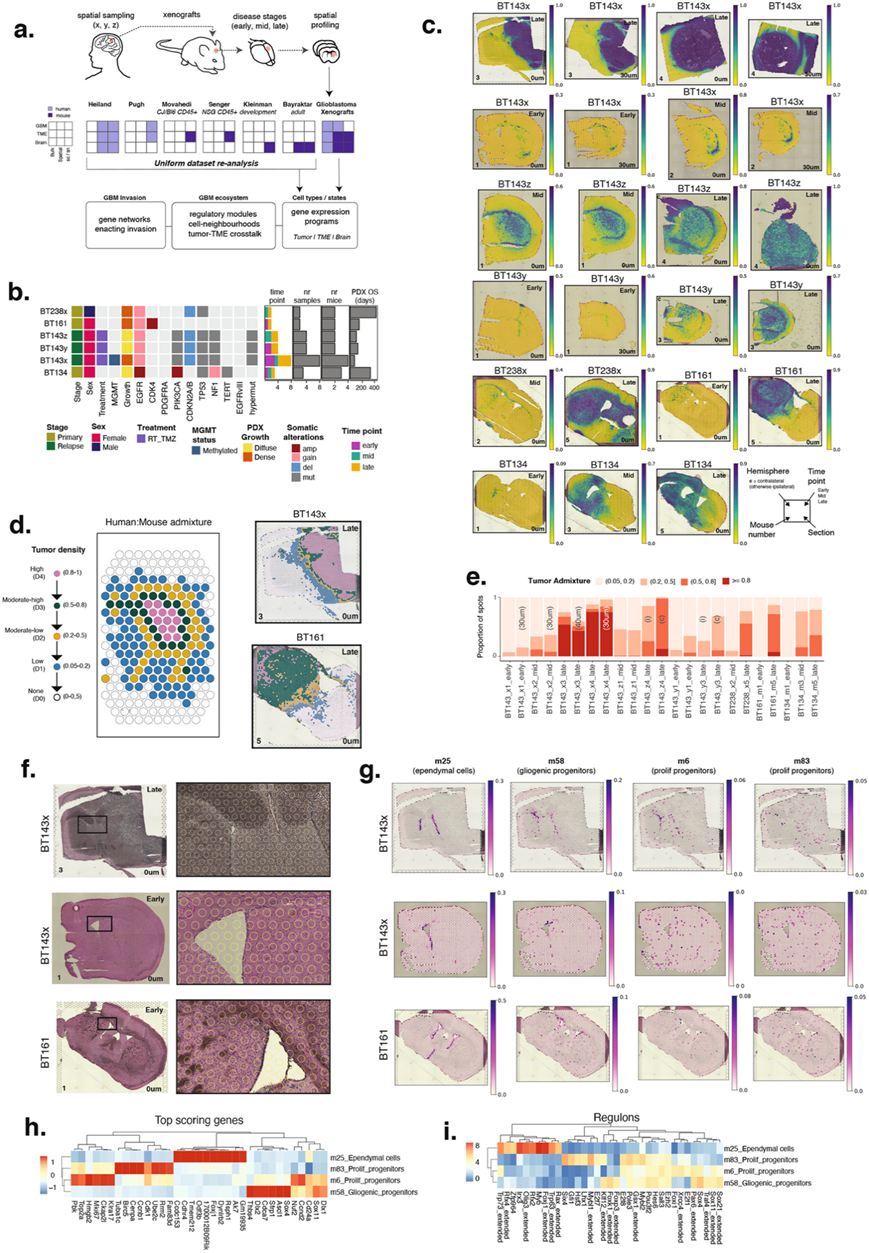
图1. 小鼠脑中的队列概览和程序特异性。
(a) 数据生成和分析的示意图。(b) 描述异种移植队列基因组特征的oncoprint。(c) 23个冠状切片的空间图显示了人类-小鼠转录组数据的比率,即基因组混合比率。(d) 肿瘤密度区示意图(左图)基于人:小鼠转录组混合物和两个选定样本(右图)分层。(e) 每个样本在D1-D4肿瘤密度范围内的斑点比例。(f) 带有斑点周长叠加的 H&E 组织学图像。(g)空间分析图。(h-i) 前10个基因的层次聚类热图和排名前10位的转录因子。
02
肿瘤程序涵盖从祖细胞状态到侵袭表型
在确定了反卷积方法的敏感性和特异性后,他们使用类似的策略表征了 15 个从头人类肿瘤细胞程序(图 2 a-d)。这包括基于通路和基于标记基因的注释、基于TF活性的相似性和空间共现性。
他们将程序分为六大主题,分别代表祖细胞状态、细胞周期、代谢、星形胶质细胞样、少突胶质细胞样和侵袭。祖细胞程序 (h5_progenitor、h13_DNArepair) 在肿瘤细胞密度高的区域 (D4) 最为普遍(图2e),以及其中一个细胞周期程序 (h7_telomere),表明循环祖细胞优先驻留在那里。OC 样程序 (h12_OC1、h14_OC2) 也显示在更密集的肿瘤区域中富集(图2e),其中h14_OC2代表 BT143x 细胞系内的遗传亚克隆。在致密的肿瘤核心之外,三个程序形成了向外肿瘤扩张的梯度,反映了以缺氧区域为中心的表型排列(h9_hypoxia位于最中心),延伸至无缺氧的肿瘤扩散更弥漫的区域 (h2_AC),最后侵入正常脑部(h11_invason)(图2 f-g),表明本研究的异种移植队列捕捉到了肿瘤生物学的这一方面,而且重要的是,通过揭示弥漫性侵袭程序(h11)扩展了以前的研究。h11_invasion程序在前沿基因集(LE_IvyGAP)(图2b)中得分很高,在早期到晚期时间点占据了 D1-D2 点的大多数,并且是唯一一个在 D1 区域显著过度表达的肿瘤程序,并且在多个个体患者中很常见(图2g)。最后,他们注意到一些程序并没有在整个队列中使用(图2d)。相反,这些程序在个体患者中普遍存在,这表明特定的肿瘤基因型可能与独特的肿瘤细胞程序库有关,而更大的异种移植队列可能会揭示更多见解。先前的研究确定 EGFR 在错误新生血管形成中发挥作用,这与肿瘤系 BT134 中血管生成肿瘤细胞程序(h3、h10)的富集一致,该肿瘤系含有高水平的 EGFR 扩增(93 个拷贝)。

图2. 肿瘤基因表达程序的特征。
(a) 根据程序基因得分(列)排序,在肿瘤基因表达程序的前 2000 个基因中显著富集的生物过程。(b) 在外部 GBM 数据集子集中针对GBM细胞状态(行)计算的肿瘤程序标记基因富集分数(列)热图。(c) 肿瘤程序之间的空间一致性热图,计算为一个程序中同时使用另一个程序(列)的点的比例(行)。(d) 15 个肿瘤程序中每个患者使用率(> 0.1)的肿瘤点(D1-D4)的比例。(e) 每个肿瘤程序使用率 > 0.01 的点比例的条形图,按肿瘤细胞密度和时间点分层。(f) 选定肿瘤程序使用情况的空间图(h2、h9 和 h11),左侧为肿瘤混合和肿瘤密度组。(g) 选定样本中富集的卡方检验结果。
03
肿瘤程序与生存和转录亚型的相关性
接下来,他们探究了这 15 个人类程序与生存差异之间的关系,预计关联可能与 GBM 转录亚型(经典、间充质、前神经)的背景相关。对于每个肿瘤程序,他们首先确定对程序身份贡献最大的基因(即得分最高的基因)(图 3 a),然后进行网络分析以根据蛋白质-蛋白质相互作用选择充当网络枢纽的子集(图 3 a),关键基因在程序之间是不同的(图 3 b)。他们使用最高评分基因和核心基因在TCGA队列中进行了生存分析,发现核心基因与所有转录亚型的生存均强相关(图3c-i)。在两项经典生存分析(根据基因集表达情况排序,比较患者的生存情况)(图3d- j)和比较患者的基因集富集时,首先根据生存结局分层(图3e-k)。有几个程序以亚型特异性的方式与生存密切相关。例如,h1_metabolism和h12_OC1的高表达与经典肿瘤的不良生存率密切相关。h1和h12基因集的 GSEA 标准化富集得分 (NES)在 TCGA 队列中高度相关,表明执行h1代谢活性(主要是胆固醇生物合成)的 OC 样细胞作为一个单位运作,共同影响与生存相关的细胞表型。间充质肿瘤按细胞周期/表观遗传 (h8_epigenetic) 和侵袭 (h11_invasion) 程序分层。h8和h11之间较低的 NES相关性表明这些程序独立影响生存。原神经肿瘤也按细胞周期 (h7_telomere) 和包含缺氧至侵袭梯度的三个程序 (h9_hypoxic、h2_AC1、h11_invasion ) 分层。TCGA 中这些程序之间的 NES 相关值遵循在异种移植 (h9-h2-h11)中观察到的相同分级共现模式,表明它们在人类患者中的空间和表型关系。
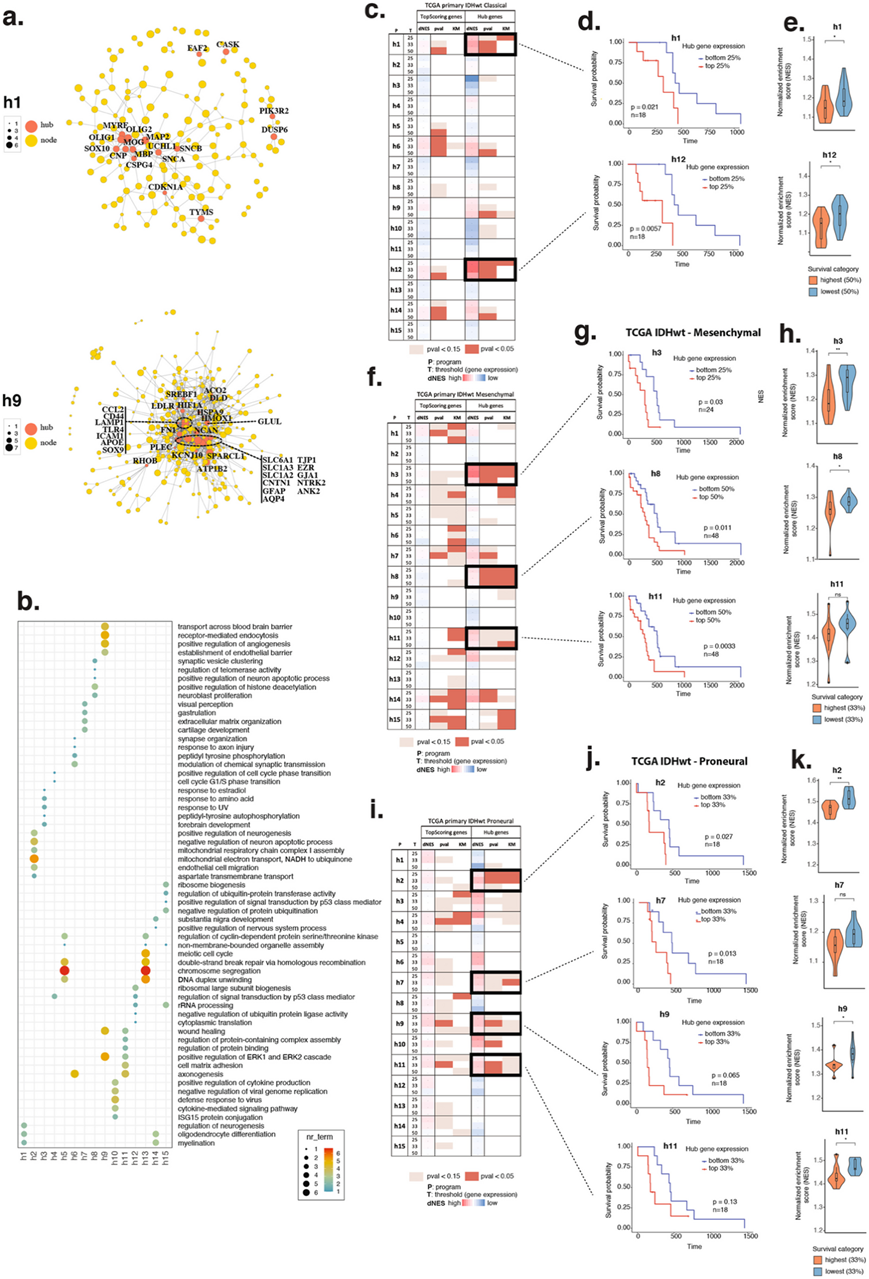
图3. 肿瘤程序的关键基因特征和亚型特异性生存关联。
(a) 选定程序的最高得分基因(黄色节点)的网络图。(b) GO分析。(c,f,i) 经典、间充质和前神经 IDHwt 原发性肿瘤的生存结果表。(d, g, j) 每种 GBM 肿瘤亚型中选定程序和阈值的 Kaplan-Meier 图。(e, h, k) 按生存率排序的患者的NES值分布。
04
微环境程序涵盖细胞类型和状态
在 90 个小鼠程序中,他们重点关注了 19 个特定于肿瘤区域或在肿瘤区域高度富集的程序,每个程序都具有动态的空间和时间动力学。根据标记基因、TF 活性和通路注释以及共定位,这些 TME 程序大致分为 11 种细胞类型和 8 种细胞活性(图 4 a-d)。当基于标记基因的注释明确且有力时,将程序标记为细胞类型;否则,标签在主题上反映了表明细胞活性相关的高度富集通路。在所有细胞系中都检测到了 TME 的所有程序(图 4 e)。
TME 程序大致沿着与肿瘤密度相对应的空间轴组织。例如,他们观察到遍布正常小鼠大脑的成熟星形胶质细胞 (m74_Astro1) 在侵袭性肿瘤前沿普遍存在,但不在密集的肿瘤区域(图4f-h)。m49_AstroRNA代表正常的星形胶质细胞活性(与 RNA 加工、增殖、谷氨酸能突触相关),在位于正常大脑和肿瘤周围的星形胶质细胞中普遍存在。然而,在肿瘤背景下,在整个肿瘤密度范围内都发生了向反应性程序 (m43_AstroReac) 的急剧状态转变,并在疾病进展的所有时间点持续(图4f)。处于这种反应状态的星形胶质细胞在与增殖、炎症反应和 ECM 组织相关的通路富集,并且与已建立的反应性特征高度匹配。总之,这些状态变化模式支持正常星形胶质细胞沿着推进的肿瘤前沿的表型选择。区域星形胶质细胞亚型或活性(m44_AstroHY)与肿瘤相关,其特征是与前体细胞增殖相关的术语的通路富集。这种看似稳态的程序在所有时间点都在下丘脑区域内高度使用(图4h),也显示出早期和持续的肿瘤富集,表明肿瘤近端星形胶质细胞转变为这种状态可能在 GBM TME 的背景下发挥重要作用。
他们观察到内皮细胞程序具有相似的区域特异性,正常脑和侵袭前沿的正常血管(m52_Endo1、m59_EndoLV )逐渐由于肿瘤密度更高、氧含量更低的区域中富含肿瘤的血管程序(m86_Endo2、m30_EndoHyp)所取代(图4 c-f)。小胶质细胞程序也弥漫在整个正常脑中(图4 f-g),对损伤部位(注射道)有早期反应,并在低密度肿瘤区域(D2-D3)内长期存在(图4 f)。其中,m7_MG1总体上更为丰富,在对损伤和抗原加工和呈递的反应中得分很高,而m77_MG2参与凋亡细胞清除,表明这些小胶质细胞亚群在肿瘤内发挥着不同的作用。 两个单核细胞衍生的巨噬细胞 (MDM) 程序迅速招募到早期病变中,而在正常大脑中则不存在(图4f-g)。 虽然这两个 MDM 程序具有大致相似的空间分布,但它们开展的活性可能与它们的微局部空间环境有关 - m40_MDM1在细胞-基质粘附、趋化性和迁移方面富集,而m84_MDM2在巨噬细胞增殖和吞噬作用方面得分很高(图4b)。在确定的多种免疫细胞活性中,他们强调了细胞毒性程序m57_Cytotoxic,它主要由 MG 和 MDM 制定(基于共定位)并定义为 D4 中最普遍的免疫活性,表明细胞毒性在更密集的区域中起着更为核心的作用(图4 f-i)。

图4. TME 基因表达程序的特征。
(a) 基于显著 GO:BP 基因集聚类的 19 个 TME 程序的基于通路的层次关系,这些基因集在每个 TME 程序的前 2000 个基因中富集(根据程序基因得分排名)(上)。基于 TF 活性得分的基于层次聚类的程序关联(下)。(b-d) 在免疫、血管和星形胶质细胞程序的前 2000 个基因中显著富集的生物过程。(e) 19 个 TME 程序中每个患者使用率 > 0.05 的点(D1 – D4)比例。(f) 条形图显示 TME 程序使用率 > 0.01 的点的比例,按肿瘤细胞密度和时间点分层。(g-i) 选定样本中的免疫、星形胶质细胞和活性程序的空间图。
05
肿瘤微环境细胞相互作用
细胞通讯对肿瘤生态系统至关重要,细胞与细胞之间的接触、肿瘤细胞外基质 (ECM) 相互作用以及分泌信号都对肿瘤的生长、适应和侵袭至关重要。为了更好地了解本文描述的神经胶质瘤和非恶性细胞如何在不同的微环境之间进行通讯,他们使用 CellChat 分析了定向配体受体 (LR) 信号传导,希望捕捉到密集区域和侵袭区域中反映 TME 组成细胞变化的不同相互作用。他们分析了所有可能的肿瘤-肿瘤、肿瘤-TME 和 TME-TME 相互作用,并按密度区域分层(图5a)。总共有 63 条通路参与了显著的相互作用,根据人类与 TME 配体和受体的方向参与情况,分为 8 个主要组(LR1-LR8)(图5b)。例如,在 TME (LR1)中,他们观察到 D1–D2 区域中的 CSF-CSF1R 信号传导涉及小胶质细胞作为主要信号接收细胞,这是基于这些程序中 CSF1R 的高分。在 LR2 中,在早期时间点,巨噬细胞和反应性星形胶质细胞之间发生了显著的 Spp1-CD44 相互作用,在后期阶段,巨噬细胞和肿瘤细胞之间也发生了显著的 Spp1-CD44 相互作用。在这种情况下,空间数据使得能够根据小鼠发送器和接收器细胞在早期时间点的空间共定位来区分小鼠与人类接收器。LR8 涉及与侵袭和干性相关的肿瘤内信号。在这一组中,CALCR 信号脱颖而出,因为 CALCRL 受体是h5_progenitor程序中得分最高的基因。CALCRL 此前已证明与神经胶质瘤细胞增殖有关,并且与神经胶质瘤预后和促进血管生成呈负相关。基于这些关联,他们推测,在 D4 中观察到的h5_progenitor程序以及肿瘤富集血管程序的富集可能与血管干细胞微环境的发育和维持有关。事实上,CALCRL 是干细胞的标志,也是其他疾病有希望的候选治疗靶点。
到目前为止,最丰富的通路涉及肿瘤和 TME 内部及之间的多向信号传导 (LR3),包括 MK 和 PTN 通路、AGRN、JAM 和 NCAM(细胞粘附)、致瘤性 NOTCH、PDGF、FGF 和血管生成性 VEGF(图 5 a-b)。他们注意到,多个 LR3 通路汇聚于 ECM(腱蛋白、层粘连蛋白、胶原蛋白、纤连蛋白)的形成和维持。层粘连蛋白和腱蛋白配体主要来自人类(LAMB2、LAMA4、TNC、TNR),而纤连蛋白配体由小鼠和人类细胞产生(图 5 d-f)。小鼠(而非人类)Fn1 在早期时间点和 D1 区域占主导地位分。由于血管程序与 Fn1(m52、m30、m86)密切相关,D1 中的主要入侵途径是沿着血管。胶原蛋白也来自肿瘤和 TME 成分,但来自肿瘤的贡献更高。人类胶原蛋白的沉积在空间上是不同的,COL6A1 在 D1-D3 中占主导地位,并且与h11_invasion程序特别相关。在 D4 中,COL9A2 和 COL9A3 更为普遍,与h1_metabolism和h7_telomere相关,表明基于密度和生态位的 ECM 沉积变化。小鼠胶原蛋白 Col4a1 和 Col4a2 在 D4 中最高,来源于肿瘤相关血管(m30_EndoHyp和m86_Vasc2中的程序得分很高)。总之,肿瘤和 TME 的细胞对肿瘤 ECM 的贡献不同,从而形成了侵袭梯度下的动态结构支架,并形成了不同肿瘤细胞微环境的基础。
在弥漫性浸润 (D1) 区域内,他们观察到通过人类受体 PTPRZ1 和 LRP1 进行的显著侵袭相关中期因子信号传导,这两个受体在h11和h2程序中得分都很高。这些受体对来自星形胶质细胞来源的中期因子配体作出反应(Mdk 在m74_Astro1和m44_AstroHY中得分很高)。Notch 信号传导也是 D1 中小鼠-人类相互作用的主要来源(LR5)(图5g -i)。Notch1 在 GBM 沿白质束侵袭中以及 GBM 细胞在血管周围微环境内的存活中都很重要。这些数据支持在 D1 区域,人类 NOTCH1 受体(在h2和h11中得分很高)可以与小鼠配体 Dlk1 和 Jag2 结合。这些配体与正常血管(m52_Vasc1中的 Dlk1)和下丘脑星形胶质细胞(m44_AstroHYP中的 Jag2)相关,突出了这些细胞程序是侵袭信号轴中不同的 TME 参与者。
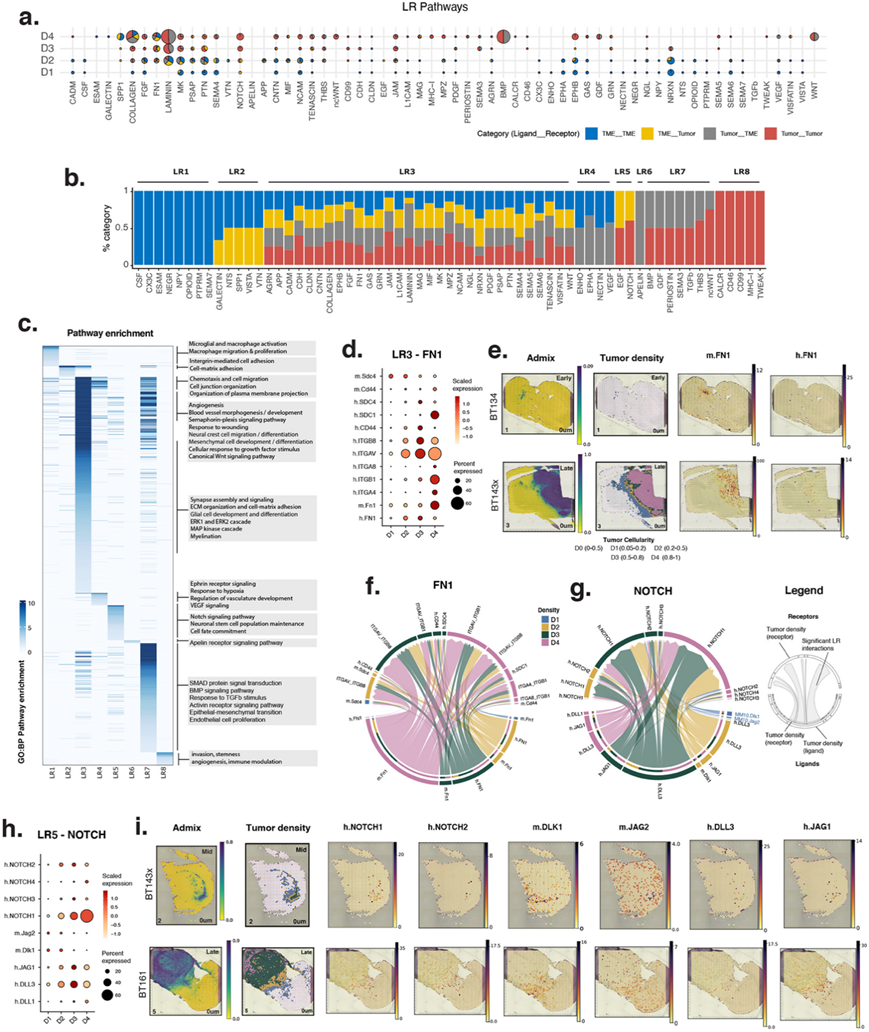
图5. 跨肿瘤密度组的配体-受体信号通讯。
(a) 显示了每个肿瘤密度组 (y 轴) 中来自 CellChat 的重要通路 (x 轴)。饼图表示人类和小鼠配体-受体相互作用类型的比例。(b) 堆积条形图表示每个通路中重要的相互作用类型比例。(c) 概述每个 LR 组中配体和受体基因之间显著富集的生物学过程。(d, h) FN和 NOTCH 通路中人类 (h.GENE) 或小鼠 (m.Gene) 配体和受体基因的表达水平。(e,i) 选定样本中人类 (h.GENE) 和小鼠( m.Gene) 基因的空间图,以肿瘤混合物和肿瘤密度为参考。(f-g) 弦图显示 FN 和 NOTCH通路的受体和配体相互作用。
06
肿瘤细胞沿不同途径侵袭的分子和结构因素
侵袭涉及肿瘤细胞沿着白质束、血管周围路径以及直接通过脑实质移动。他们尝试通过小鼠脑程序与肿瘤侵袭的关联来识别和描述这些路径,然后进行 CellChat 分析和差异表达分析。他们首先评估了所有 90 个小鼠脑程序在 D1 点中的过度代表性,期望看到与侵袭相关的细胞类型或解剖区域的富集(图6 a-b)。事实上,白质 (WM) 相关程序与 D1 肿瘤区域的关联性最高。因此,他们选择了小鼠 WM 程序使用率高的 D1 点作为沿白质束移动的人类肿瘤细胞的代表。同样,他们观察到尾壳核程序(m1、m73)的使用在 D1 区域也显著富集(图6a-b)。进一步分析表明,许多尾壳核点也使用血管程序;因此,他们根据四种血管程序的共同使用情况进一步对尾壳核点进行分层。该策略将沿每种血管类型(血管周围路径)行进的 D1 肿瘤细胞与直接穿过脑实质(即没有血管的尾壳核点)移动的肿瘤细胞区分开来。NPC 样和 OPC 样状态在沿白质束行进的 D1 肿瘤细胞中更常见,而脑实质内的肿瘤细胞则偏向 MES 和 AC 样状态(图6c)。因此,尽管h11和h2侵袭程序具有高度可塑性,但侵袭前沿的细胞状态决策反映了对特定路径局部细胞环境的适应。
他们对6 组 D1 点进行了另一次 CellChat 分析:白质 (WM)、实质 (Par) 和血管周围路线(m52、m30、m86、m59)(图 6d)。胶原蛋白、纤连蛋白和层粘连蛋白相互作用显著且普遍存在于所有路线上(图 6d)。人类整合素受体与各种小鼠胶原蛋白的相互作用主要按脉管系统程序分层,这表明肿瘤细胞利用 TME 内现有的胶原蛋白支架进行运动。一个显着的例外涉及实质和m52区域中的人类 COL6A1 相互作用,这表明肿瘤细胞特别需要并分泌这种 ECM 成分(图 6e)。类似地,肿瘤细胞不仅与多种小鼠层粘连蛋白结合(图 6e),还为 ECM 贡献了一组精选的配体(实质中的 LAMA5 和脉管系统中的 LAMB2)(图 6e)。一种层粘连蛋白是 WM 途径所特有的(小鼠 Lama2)(图 6e)。该基因在程序m71 (新形成的少突胶质细胞)中排名很高,表明这种少突胶质细胞亚型有助于迁移支架。总之,这些结果强调了 TME 衍生的 ECM 可以区分侵袭途径,值得注意的是,一组区域特异性成分来自肿瘤细胞本身——表明这些分子对胶质瘤细胞沿不同行进路线移动的重要性。
CellChat 分析还揭示了几种特定于实质的信号相互作用,包括 NRXN、CD99、PSAP、PTN、MK、CNTN 和 NOTCH(图6 d)。其中,他们重点分析了肿瘤(NRXN1)与神经元衍生的突触粘附分子神经连接蛋白(Nlgn1、Nlgn2、Nlgn3)的相互作用(图6 d-e)。这种先前描述的有丝分裂原信号轴将兴奋性神经元活性转向肿瘤生长,表明活跃的神经元在胶质母细胞瘤通过实质侵袭中发挥作用。此外,他们还进一步观察到肿瘤细胞通过NRCAM(神经元细胞粘附分子)受体和小鼠Cntn1配体(一种在胆碱能神经元和GABA能神经元中得分很高的基因)之间的相互作用,这表明这些神经元可能在侵袭过程中发挥除兴奋性活性之外的其他作用(图6e)。
最后,他们结合了所有针对 D1 的分析的数据,包括完整的 CellChat 分析(图5a)、针对侵袭途径的 CellChat 分析以及上述两个差异表达分析。将得到的基因与肿瘤程序相交,发现有 27 个基因重叠,这些基因也是预后侵袭相关程序h2和h11中得分最高的基因或网络枢纽基因(图6f)。其中 9 个是h2(PTN、PTPRZ1、LRP1、NRXN1、VEGFA)或h11(SDC3、COL6A1、NCAM1、NOTCH2)中的枢纽基因,而 2 个基因是两者中的枢纽基因(FN1、APP)(图6f)。此外,在 TCGA 队列中,其中三个基因与前神经亚型肿瘤的不良生存结果独立相关(TUBA1A、NRCAM、LGALS1)(图6g)。 NRCAM 是一种神经粘附分子,参与神经发育过程中的细胞增殖、轴突生长和突触形成,此前有研究表明它在脑肿瘤中过表达。TUBA1A 是一种微管亚基,在神经元迁移中具有效应功能,与神经发育缺陷有因果关系。LGALS1(半乳糖凝集素-1)通过调节细胞-细胞和细胞-基质相互作用在 GBM 侵袭中发挥作用,还能促进免疫抑制性 TME。另外两个基因与 TCGA 间充质肿瘤(COL6A1、FN1)的不良生存率独立且显著相关(图6h)。这两个基因之前均与 CD133+ 胶质瘤癌症干细胞相对于分化的胶质母细胞瘤细胞在 ECM 中的沉积增加有关,并且在患者样本的血管周围和 PAN 区域中观察到了 COL6A1。
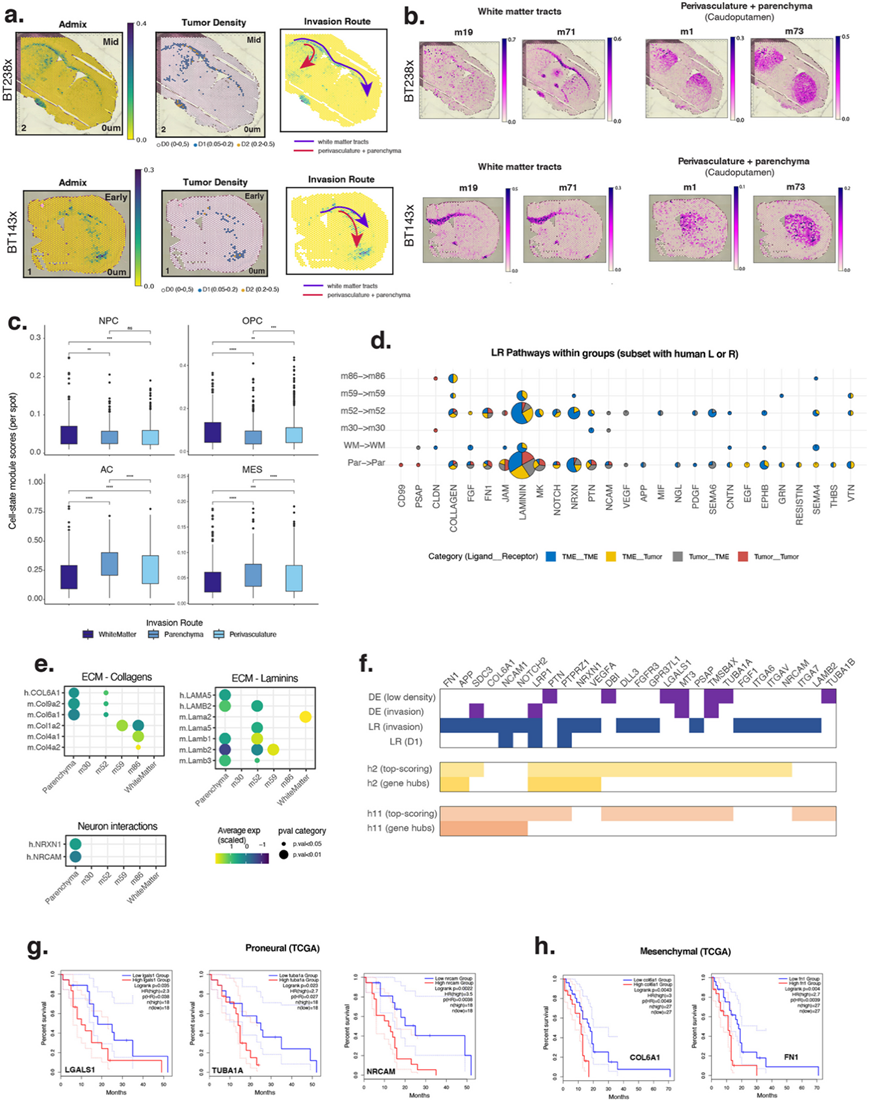
图6. 与GBM 侵袭途径相关的基因特征和信号通路。
(a-b) 程序使用情况的空间图(右图),分为白质(m19 或 m71)或尾壳核(m1 和 m73),包括血管和实质侵袭途径,以及混合物、肿瘤密度和感兴趣的侵袭途径作为参考(左图)。(c) 白质、实质或血管周围侵袭途径中每个 GBM 细胞状态的每个点的模块得分的箱线图。(d) 对于具有至少一种人类配体或受体的通路子集,每个 D1 类别组(y轴)显示了来自 CellChat 的显著通路(x轴)。饼图显示了人类和小鼠配体-受体相互作用类型的比例。(e) 参与白质、实质或血管周围 (m30、m52、m59、m86) 侵袭途径中 ECM和神经元相互作用的选定基因的表达水平。(f) 基于多项评估的侵袭相关基因总结。(g-h) Kaplan-Meier 生存曲线。
+ + + + + + + + + + +
结 论
本项研究将一种新颖的计算工作流程应用于时空分析的 GBM 异种移植队列,利用区分人类肿瘤和小鼠 TME 的能力来克服弥漫性侵袭分析中的先前局限性。该分析方法基于无监督反卷积,可在完整的 GBM 生态系统中执行无参考的细胞类型和细胞活性分析。本研究提供了 15 个肿瘤细胞程序的综合目录,这些程序设置在 90 个小鼠大脑和 TME 细胞类型、细胞活性和解剖结构的时空背景下。与侵袭相关的不同肿瘤程序与血管周围、白质和实质侵袭的途径一致。此外,作为程序网络中心的基因子模块对 GBM 患者具有高度的预后性。
+ + + + +



