

 English
English文献解读|iScience(5.8):血液代谢组学和转录组学特征根据疾病严重程度对多发性硬化症患者亚组进行分层
✦ +
+
论文ID
原名:Blood metabolomic and transcriptomic signatures stratify patient subgroups in multiple sclerosis according to disease severity
译名:血液代谢组学和转录组学特征根据疾病严重程度对多发性硬化症患者亚组进行分层
期刊:iScience
影响因子:5.8
发表时间:2024.02.15
DOI号:10.1016/j.isci.2024.109225
背 景
多发性硬化症 (MS) 是一种自身免疫性疾病,同时具有炎症和神经退行性成分,影响全球约 250 万人。大约 85% 的多发性硬化症患者诊断患有复发缓解型疾病 (RRMS),其特征是可逆或接近可逆的神经功能损伤(复发)期。RRMS 的特点是全身性自身炎症,导致神经元脱髓鞘、少突胶质细胞和神经轴突损伤,这些损伤可能与残疾增加有关。尽管有证据支持根据 MS 疾病严重程度进行代谢组学变化,但没有基于血液的生物标志物能够区分RRMS患者和继发性进行性多发性硬化症 (SPMS)患者。
实验设计
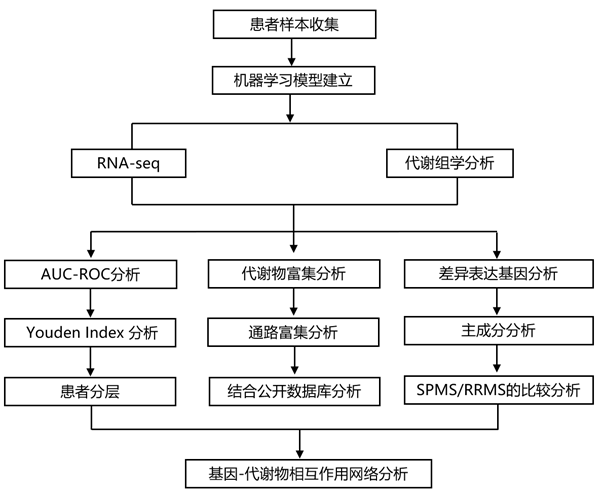
结 果
01

血清代谢物可以在多发性硬化症患者亚组以及健康和疾病对照之间分
研究团队使用NMR血清代谢组学平台分析了复发-缓解型多发性硬化症(RRMS)、继发性进展型多发性硬化症(SPMS)、健康供体(hc)和视神经脊髓炎[对照(DC),一种自身抗体介导的疾病,具有某些症状,可能误诊为多发性硬化症]患者的血清。使用七种机器学习 (ML) 模型对各组之间的代谢组数据进行比较(图 S1)。代谢组数据的 ML 分析还能够对 SPMS 与 RRMS 患者进行分层。总体而言,LR 和随机森林 (RF) 模型在预测 SPMS 病例时表现最佳,正确识别了 29 名患者中的 26 名 (89.7%)(图 1 A)。而增强 LR 对于 RRMS 病例表现更好,具有出色的特异性 (0.961) 和 93.67% 的准确度。
图S1. 使用机器学习模型进行代谢组学和转录组学分析。
为了得出可以区分 SPMS 和 RRMS 患者的特征,他们对所有分析的特征进行了比较(图 1 B)。三个或更多模型中包含了 36 项指标,包括代谢物、血脂和临床人口统计信息。最重要的特征包括谷氨酰胺 (Gln) 以及临床特征年龄和扩展残疾状态评分(EDSS),这些特征出现在所有七个模型以及稀疏偏最小二乘判别分析(sPLS-DA)模型中(图 1B)。亚油酸(LA)、中低密度脂蛋白胆固醇酯(M-LDL-CE)、小低密度脂蛋白游离胆固醇(S-LDL-FC)、乙酰乙酸、饱和脂肪酸(SFA)、omega-6脂肪酸(%)、单不饱和脂肪酸和多不饱和脂肪酸(MUFA/PUFA %)和超大极低密度脂蛋白游离胆固醇(XL-VLDL-FC %)通过6个ML模型和/或sPLS-DA进行鉴定(图 1B)。此外,糖酵解相关代谢物(乳酸、葡萄糖、丙酮酸和柠檬酸)、氨基酸(缬氨酸、酪氨酸、组氨酸和丙氨酸)和鞘磷脂在三种或更多ML模型中都有表现(图 1B)。
正如预期的那样,所有模型都确定了与严重程度和进展相关的年龄和 EDSS(图1B)。使用LR与相互作用(LR + I)模型进一步检测年龄和EDSS的影响(图1A)。虽然排除年龄和EDSS会降低模型准确率(88.9%-80.3%)和AUC ROC(0.936-0.844),但单独使用代谢物仍能对SPMS和RRMS患者进行分类,效果良好。通过三种或三种以上ML模型(排除年龄和EDSS)鉴定的代谢组学特征对SPMS和RRMS患者进行分类的能力得到了sPLS-DA的证实(图1C)。曲线下面积——受试者工作特征曲线(AUC ROC) 分析显示,在区分SPMS患者与RRMS患者时,增强型LR表现最好(图1D)。
为了开发一种基于血清代谢组学对SPMS和RRMS患者进行分层的可靠方法,他们计算了在>4 ML模型中鉴定出的每种血清代谢物的最佳临界值。选取Youden Index >0.5的特征,建立基于积分的患者群体分层系统。自动评分模型确定了前五个特征(胆碱、谷氨酰胺、饱和脂肪酸、乙酰乙酸酯和鞘磷脂)的最佳临界值(图1F)。最终模型的性能在测试集(30%的患者)中得到证实,该测试集将RRMS与SPMS分层,AUC ROC为0.9464(图1F)。该分析表明,血清代谢物可以用来开发一种可靠的方法来对患者进行分层,这种方法可能比EDSS和年龄等严重程度的临床标志物更有效。
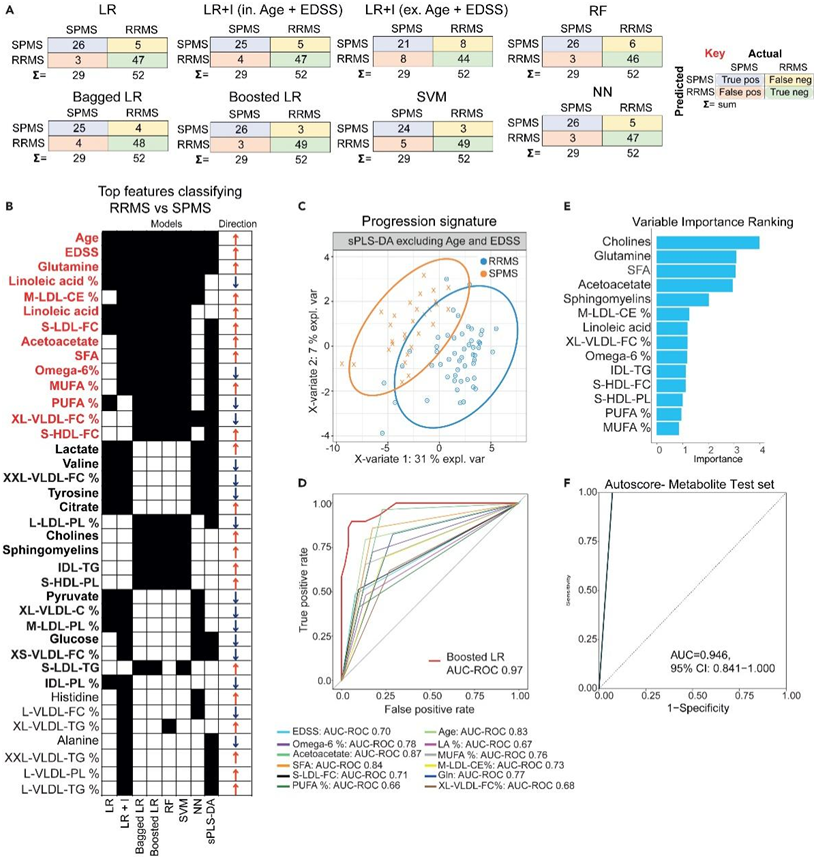
图1. 血清代谢组学可以对 SPMS 与 RRMS 患者进行分层。
(A) 混淆矩阵显示每个模型的正确分类(蓝色方块)和错误分类(绿色方块)的数量。(B) 每个机器学习模型选择的代谢物的比较。(C) sPLS-DA 图用于验证 (B) 中由 ≥ 3 个模型鉴定的代谢物中的代谢组学特征。(D) 由 ≥ 6 个模型和性能最佳的机器学习模型确定的前 14 种代谢物和/或临床特征的AUC-ROC。(E-F) 使用 Youden Index 分析中的代谢特征,以确定最能对 SPMS 与 RRMS 患者进行分层的特征。
02
继发性进行性多发性硬化症患者的葡萄糖、脂质和氨基酸代谢发生改变
为了评估ML模型鉴定的代谢物是否表明RRMS与SPMS患者之间代谢过程的差异,他们进行了代谢物富集分析(图2A-B)。与RRMS相比,SPMS患者与蛋白质合成、氨基酸、脂质代谢和细胞呼吸相关的代谢通路富集,包括“酮体合成与降解”/“酮体代谢”、“甘油脂代谢”、“糖异生”和其他与细胞呼吸相关的通路如糖酵解、“丙酮酸代谢”和“柠檬酸循环”(图2A-B)。网络分析显示,这些通路是相关的(图2B),并支持SPMS患者与以糖异生和生酮增加为特征的RRMS患者相比,细胞呼吸的潜在变化。通过比较血清代谢物浓度支持了这一观察结果:与RRMS相比,SPMS患者的糖异生相关代谢物(乳酸和谷氨酰胺)、酮体(乙酰乙酸酯和bOHbutyrate)和柠檬酸盐显著升高(图2C-2E),而糖酵解相关代谢物(丙氨酸和丙酮酸)降低(图2F)。与RRMS患者相比,SPMS患者的脂质(如甘油三酯、胆碱)和脂肪酸代谢物也出现失调(图2G)。总的来说,这些数据显示了SPMS和RRMS患者血清代谢特征的复杂变化。
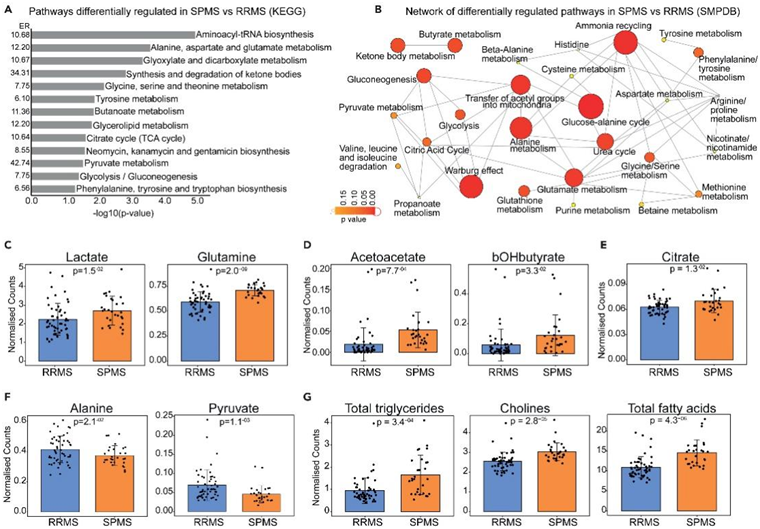
图2. SPMS 与 RRMS 代谢组特征的代谢物富集分析 (MSEA) 。
(A) 使用 KEGG 数据库进行富集分析。(B) 来自 SMPDB 数据库的代谢通路网络。(C–G) 条形图显示 RRMS (蓝色)与 SPMS(橙色)患者血清中代谢物的相对表达。
03
继发进行性多发性硬化症患者的全血转录组中反映了差异代谢物表达
为了评估血清代谢组的变化是否得到更广泛的反映,他们对一部分患者进行了全血转录组分析(RNA-seq)。比较 MS 患者组的差异表达分析确定了 1052 个差异表达基因 (DEG),这些基因能够对 RRMS 和SPMS患者进行聚类(图 3 A)。其中,948 个基因上调,104 个基因下调(图 3 B)。SPMS 与 RRMS 中上调和下调基因的通路富集分析还确定了与细胞代谢相关的通路,包括“RNA 代谢”、“细胞对应激的反应”、“脂质代谢”和“细胞呼吸”(图3C-D,图S3A-E)。总共有 215 个 DEG 与上述代谢通路相关,这包括以下基因:核呼吸因子 1 (NRF1)、CC 趋化因子受体 5 型 (CCR5)、谷氨酸草酰乙酸转氨酶 2 (GOT2)、线粒体调节蛋白 (LINC00116/MTLN) 和 O-唾液酸糖蛋白内肽酶 (OSGEP)。与 RRMS 相比,SPMS 患者的免疫激活通路(包括“TNFA 信号传导”和“免疫系统中的细胞因子信号传导”以及“免疫效应过程的调节”)也有差异富集(图3D-E,S3A-F)。“细胞呼吸通路”的网络分析揭示了与“肌萎缩侧索硬化症”的关联(图S3 G),支持 SPMS 神经退行性过程中细胞代谢失调的作用。
最后,当与一个独立的基因表达数据集进行比较时,也确定了SPMS患者与RRMS患者相比富集的许多代谢和免疫相关通路,该数据集将SPMS患者的全血、淋巴细胞、髓细胞和少突胶质细胞前体细胞与RRMS患者样本进行比较(图3E)。总之,这些结果表明,与RRMS患者相比,SPMS患者的代谢通路发生了系统性改变,主要集中在RNA生物合成、脂质代谢和细胞呼吸通路。
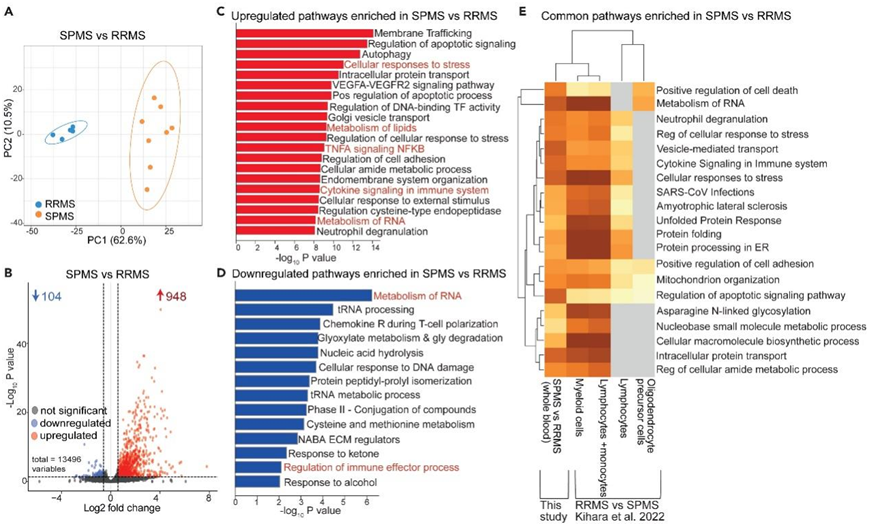
图3. SPMS和RRMS患者之间的RNA代谢、细胞对应激的反应、免疫效应反应、细胞呼吸、脂质代谢和tRNA加工通路失调。
(A) 对所有 1052 个 DEG 进行主成分分析(PCA),将 SPMS 患者(橙色)与 RRMS 患者(蓝色)进行聚类。(B) 火山图显示差异表达基因 (DEG) 变化。(C-D) 通过 Metascape 进行通路富集分析。(E) 使用独立数据集验证基因表达通路。
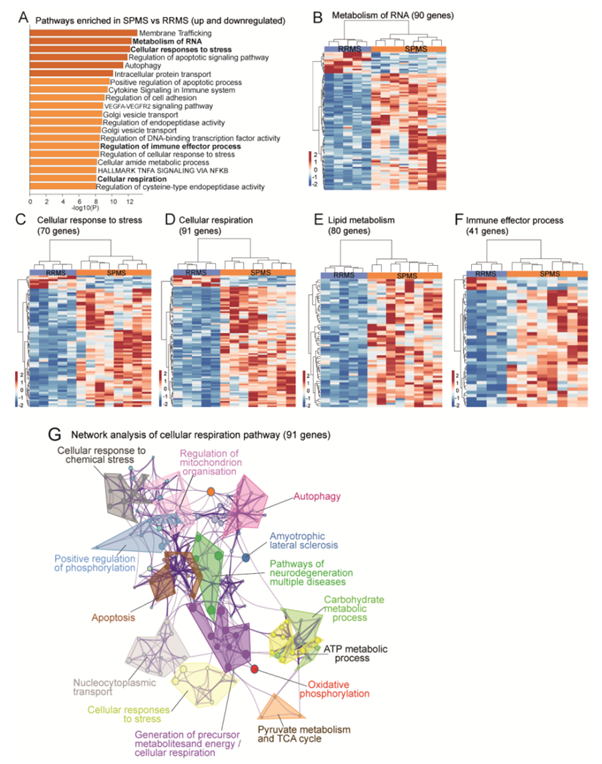
图S3. SPMS与RRMS的通路失调。
(A)通过metscape分析所有1052个deg的通路富集分析,以确定失调的通路。(B-F) 不同通路中标准化基因计数的热图。(G)网络图显示了细胞呼吸通路中与基因显著上调和下调相关的通路。
04
基因-代谢物相互作用网络提出了多发性硬化症严重程度代谢转换背后的潜在机制
他们使用相关性分析进一步探讨了匹配样品中代谢物与DEG之间的关系。在细胞呼吸、免疫效应反应通路的调节、脂质代谢和RNA代谢中,代谢产物和DEG之间存在多重显著相关性(图S4A-D)。这些相关性大多是正的,大量的相关性表明,从转录组到代谢组水平,代谢产物和与细胞呼吸和代谢相关的基因之间存在协调。
为了进一步整合代谢组学和转录组学数据,他们对所有1052个deg和SPMS代谢组学特征进行了网络分析。在最终的网络中,发现26个上调基因和4个下调基因与代谢组学特征中的8种代谢物(肌酐、柠檬酸、丙酮酸、乳酸、苯丙氨酸、酪氨酸、甘氨酸和亚油酸)相互作用(图4)。虽然脂质不能包含在网络内,但上调基因 SCD5(硬脂酰辅酶 A 去饱和酶 5)、PLA2G12A(磷脂酶 A2 第 XIIA 组)、CARM1(辅激活剂相关精氨酸甲基转移酶 1)和 SLC25A1(可溶性载体家族 25 成员 1)均在网络内脂质通路中。下调的基因 GOT2 与多种代谢通路相关,包括氨基酸代谢、氨酰 tRNA 生物合成、脂质代谢、丙酮酸代谢、糖异生和酮生成。
总而言之,这个基因-代谢物相互作用网络展示了关键基因和代谢物之间的相互作用,这可以突出与 MS 严重程度相关的分子过程。
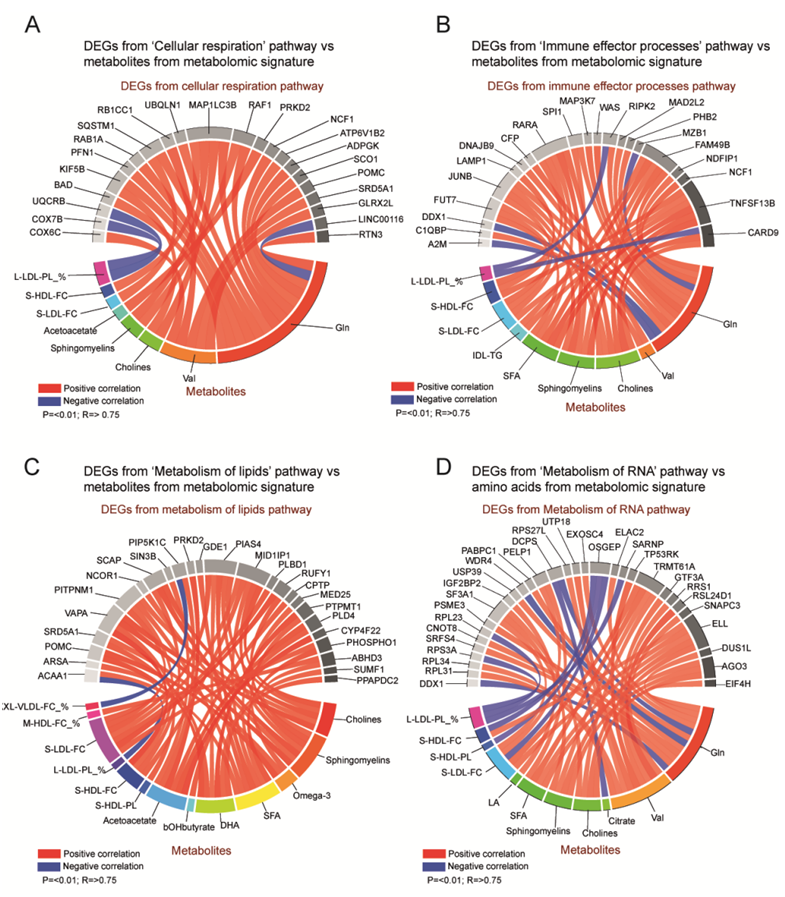
图S4. deg与代谢物的相关性。
(A)细胞呼吸通路中的DEG与代谢组学标志物浓度之间存在显著相关性。(B)“免疫效应过程调节”通路中的deg与代谢组学标记物浓度显著相关。(C)“脂质代谢”通路中的deg与代谢组学标记物浓度之间存在显著相关性。(D) RNA代谢通路中的deg与代谢组学标记物浓度之间存在显著相关性。
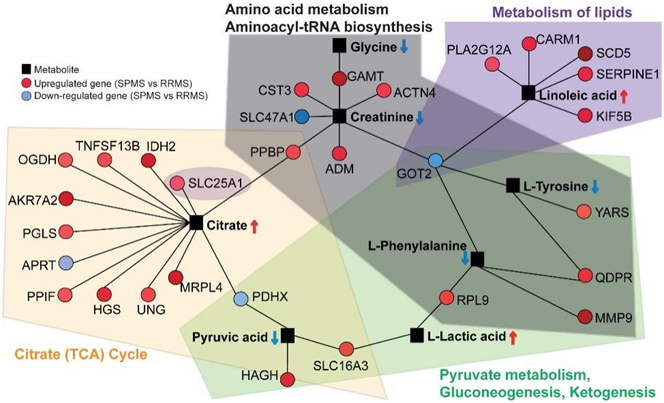
图4. 基因-代谢物相互作用网络。
+ + + + + + + + + + +
结 论
本项研究对血清代谢组数据进行机器学习模型分析,以高精度对SPMS和RRMS 患者进行分层,并开发了对 MS 患者子集进行分层的推定评分。SPMS 与 RRMS 患者之间最差异表达的代谢物包括脂质和脂肪酸、富含与细胞呼吸相关通路的代谢物,特别是乳酸、谷氨酰胺(糖异生相关)、乙酰乙酸和丁酸(酮体)升高,以及丙氨酸和丙酮酸(糖酵解相关)降低。全血转录组中重现了血清代谢组的变化,SPMS 患者的细胞呼吸通路中也富集了差异表达基因。最终的基因-代谢物相互作用网络证明了 SPMS 中从糖酵解到增加糖异生和酮生成的潜在代谢转变,表明代谢应激可能触发应激反应通路和随后的神经变性。
+ + + + +



