

 English
English文献解读|Nat Commun(16.6):TRIM40通过破坏肠道屏障完整性驱动炎症性肠病的发生
✦ +
+
论文ID
原名:TRIM40 is a pathogenic driver of inflammatory bowel disease subverting intestinal barrier integrity
译名:TRIM40通过破坏肠道屏障完整性驱动炎症性肠病的发生
期刊:Nature Communications
影响因子:16.6
发表时间:2023.2.9
DOI号:10.1038/s41467-023-36424-0.
背 景
肠道上皮的完整能够防止肠道微生物群或有害外源性因素易位,从而维持免疫稳态,破坏肠道保护屏障会导致 炎症性肠病(IBD)的发作和进展。尽管IBD的主要特征是与炎症相关的特定基因表达模式、组织完整性、细胞粘附和屏障功能的改变,而这些变化是IBD的致病因素还是发病结果尚不清楚。此外,这些变化的分子机制及它们引发IBD发展阶段尚待阐明。肌动蛋白细胞骨架在维持肠道屏障完整性方面起着关键作用,由细胞骨架结构扰动诱导的上皮功能障碍导致紧密连接或粘附连接(AJ)的丧失能够增加肠道通透性。Rho相关含线圈的蛋白激酶1(ROCK1)是肌动蛋白聚合和稳定的必需激酶,在肌动蛋白动力学和细胞迁移中起着重要作用。然而目前的研究对ROCK1的表达或稳定性调节的具体机制知之甚少。在这里,作者将TRIM40确定为IBD的致病驱动因素,其直接靶向ROCK1进行降解,导致肌动蛋白细胞骨架稳定所需的下游信号转导事件的破坏,导致上皮屏障功能衰竭,并最终导致IBD的发展。这项研究提供了概念证明,TRIM40的表达是肠道慢性炎症的主要病理驱动因素,因此可能是限制IBD起始和发展的潜在生物标志物或治疗靶点。
实验设计
结 果
01

TRIM40上调通过改变上皮细胞形态和骨架驱动IBD发生。
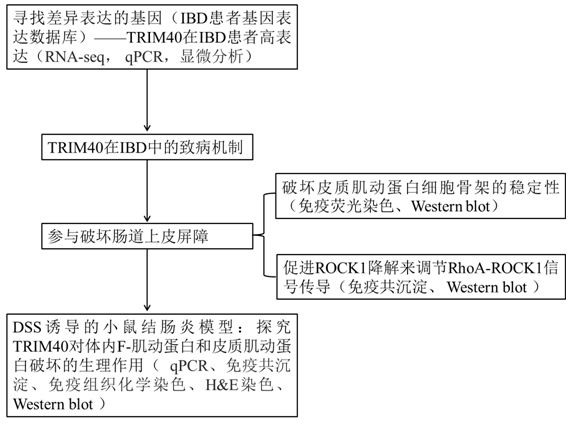
为了确定驱动IBD潜在致病因素,作者访问了IBD患者直肠活检样本的公共基因表达数据集,包括溃疡性结肠炎(UC)和克罗恩病(CD)患者。通过评估与诱导IBD相关的特征基因(包括与细胞因子、补体系统、组织完整性、屏障功能和抗菌肽相关的基因),发现 UC 和 CD 患者中 TRIM40 和 TRIM69 的基因表达水平相对于健康对照组显著增加(图1A)。与TRIM69 不同,TRIM40 的基因表达在大多数人体组织和人源性细胞系中几乎检测不到,包括结肠上皮细胞(HT-29、HCT116 和 Caco-2),这与TRIM31在肠道中高表达形成鲜明对比(图1B);此外,TRIM40在IBD样品中的表达远高于非IBD健康人肠道组织(图1B)。作者分析了来自IBD患者直肠和回肠区域的另外三个转录组测序(RNA-seq)数据集,结果表明,UC和CD患者直肠样本中的TRIM40显著增加,但在回肠组织中没有(图1C)。在UC患者的直肠粘膜(疾病特别受累部位)中也观察到异常的TRIM40过表达(图1D)。使用来自UC和CD患者炎症区域的直肠活检标本证实了这一现象(图1E)。
为了评估IBD患者的TRIM40蛋白的表达水平是否升高,我们使用人TRIM40特异性抗体进行了免疫组织化学染色,与非IBD的对照组不同,TRIM40蛋白在UC和CD的结肠组织中显著着色(图1F)。作者在人结肠上皮细胞系HT-29中进行RNA-seq和微阵列分析来探索TRIM40过表达对RNA转录本全局特性的影响,HT-29稳定地过表达N末端Myc标记的TRIM40(Myc-TRIM40)。值得注意的是,TRIM40过表达的HT-29细胞的转录组图谱显示出IBD特征基因的实质性变化,包括细胞骨架相关的基因和干扰素刺激基因(ISG),这一结果也通过定量PCR(qPCR)测定得到证实(图1G,H)。TRIM40主要在细胞质中表现出丝状图案,在细胞核内检测到微弱的信号, GFP标记的TRIM40(GFP-TRIM40)的过表达与异常形态变化相吻合,包括细胞间排斥,相邻细胞之间的显着间隙,以及最终的扩散(图1I 右);在形成紧密簇的对照HT-29细胞中未观察到这些形态变化(图1I 左)。TRIM40过表达的HT-29细胞之间的平均边距也明显大于对照组HT-29细胞(图1J)。这些结果表明,表观遗传沉默的TRIM40的高表达可能作为潜在的内在线索,导致结肠上皮细胞细胞骨架结构的扰动。
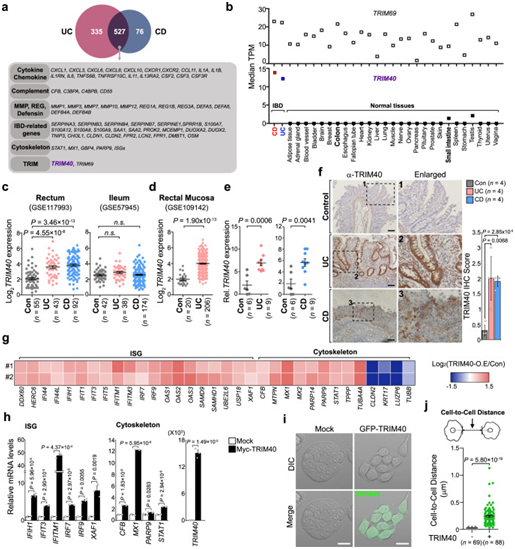
图1:上调的TRIM40通过改变细胞形态和骨架促进IBD 发生。
(A) 维恩图说明了UC和CD患者以及健康对照组之间重叠的差异表达基因。(B) 基因型-组织表达项目中IBD(GSE117993)RNA-seq数据与正常组织TRIM40或TRIM69表达的对比分析。(C)、(D) UC、CD和正常样本(GSE109142、GSE117993和GSE57945)在直肠和回肠(C)或直肠粘膜(D)中TRIM40表达的比较。(E) qPCR实验比较IBD患者与正常组织TRIM40 mRNA水平。(F) IHC 分析对照组、UC或CD 患者TRIM40的表达。(G) 热图显示了RNA-seq的指示基因在对照载体或表达Myc-TRIM40的HT-29细胞中的相对表达。(H) qPCR显示对照载体或表达 Myc-TRIM40的HT-29细胞中指示基因的相对mRNA水平。(I),(J) 显微分析显示对照组或表达GFP-TRIM40的HT-29细胞的形态改变。
02
TRIM40通过破坏皮质肌动蛋白细胞骨架的稳定性来破坏上皮完整性。
由于肌动蛋白细胞骨架是调节细胞间粘附的核心,作者进一步探讨了TRIM40表达对肌动蛋白动力学的影响。对照组HT-29细胞有与质膜结合的薄肌动蛋白网络层,显示出组织良好的皮质结构(图2A);然而,过表达TRIM40的细胞表现出皮质F-肌动蛋白的回缩和碎裂,以及F-肌动蛋白在细胞内接触位点的不规则积累和部分TRIM40的丝状拉伸(图2A)。预粘附连接(AJ)通常被观察到为与细胞-细胞接触区域对齐的点状结构,通过促进AJ组分募集到AJ区域来驱动相邻细胞之间的膜闭合,形成细胞-细胞粘附拉链。值得注意的是,尽管细胞间边界存在F-肌动蛋白点,但TRIM40过表达细胞未能形成粘附拉链(图2A)。这些细胞特异性地含有主要AJ成分(CD44和E-钙粘蛋白)的不连续簇,它们位于表面膜或细胞间区域,而不是被募集到AJ区域(图2B)。作者观察到大多数(>80~90%)的黏着蛋白位于局灶性粘连附近,在正常条件下,少数细胞在细胞-细胞接触区域显示黏着蛋白染色。然而,在表达TRIM40的细胞在细胞-细胞接触位点甚至无法检测到黏着蛋白的微弱荧光信号(图2B)。AJ成分的这种分布和相邻细胞之间交叉的丧失与对照组细胞不同,对照细胞的特征是沿着绝大多数侧细胞边界的密集,连续的AJ区域,没有空隙(图2B)。为了验证TRIM40有助于F-肌动蛋白破坏的可能性,从对照和TRIM40过表达细胞中分离出富集的G-和F-肌动蛋白部分。与对照细胞相比,TRIM40过表达细胞中可溶性G-肌动蛋白的量显著增加;值得注意的是,F-肌动蛋白减少了类似的量(图2C)。
作者设计了一个用于多西环素诱导的TRIM40表达的Tet-On系统,能够以受控的方式过表达TRIM40,并准确监测TRIM40在上皮细胞中过表达的具体影响。在多西环素处理后,HT-29细胞中的p-ERM水平显著下降(图2D,E),而TRIM40的表达最大化。此外,在过表达TRIM40的细胞中,上皮膜完整性也被破坏,如测量所示,上皮细胞中的电阻抑制为>80%(图2F)。作者的研究结果表明,表观遗传抑制的TRIM40的过度表达会导致病理功能的增加,这与一系列影响上皮细胞完整性的分子过程有关,包括ERM磷酸化、潜在的皮质F-肌动蛋白形成和稳定。
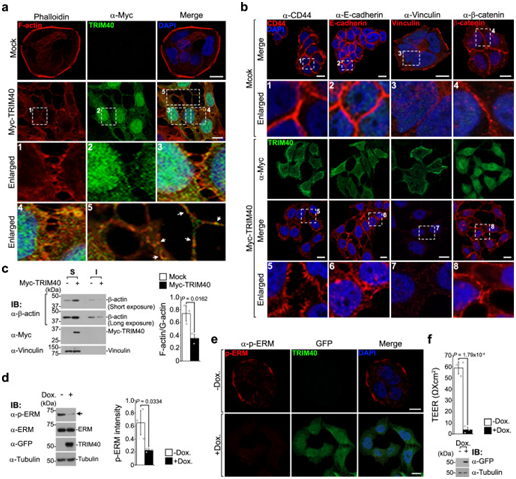
图2:TRIM40 通过破坏皮质肌动蛋白细胞骨架的稳定来破坏上皮完整性。
(A)、(B) 对照组细胞与表达Myc-TRIM29载体的HT-40细胞的共聚焦荧光图像。(C) 免疫印迹显示对照组细胞与表达Myc-TRIM29载体的HT-40细胞的可溶性(S)和不溶性(I)组分中的β-肌动蛋白水平。(D) 免疫印迹显示,使用多西环素诱导系统暂时过表达GFP-TRIM29的HT-40细胞中p-ERM水平降低。(E) 多西环素诱导的GFP-TRIM40过表达细胞中GFP-TRIM40 (绿色)和磷酸化ERM (p-ERM,红色)的共聚焦荧光图像。(F) 检测经多西环素处理的过表达GFP-TRIM40的Caco-2细胞的跨上皮电阻(TEER)值。
03
TRIM40通过促进ROCK1降解来调节RhoA-ROCK1信号传导。
Rho家族的GTPases-Rac1、Cdc42和RhoA是最常见的分子开关,能够激活两种主要的激酶PAK和ROCK1(图3A)。PAK和ROCK1的激活触发下游关键效应蛋白LIMK1、MLC2和ERM的磷酸化,最终促进F-肌动蛋白和皮质肌动蛋白的形成和稳定(图3A)。在TRIM40过表达HT-40的TRIM29上皮细胞中,ROCK1蛋白水平显著降低,但mRNA水平没有降低(图3B);然而并未在ROCK1的上游或下游蛋白质RhoA,LIMK1,MLC2,cofilin1(CFL1)和profilin1(PFL1)观察到类似的结果(图3B)。TRIM40过表达阻断了LIMK1和MLC2的磷酸化,它们是ROCK1的已知底物,此外还观察到对ERM磷酸化的抑制作用(图3B)。最终,抑制LIMK1磷酸化导致CFL1磷酸化的破坏(图3B)。使用正常的结肠上皮细胞系FHC证实了这一点,表明TRIM40过表达明显降解ROCK1,随后破坏FHC细胞中的CFL1磷酸化(图3C)。由于TRIM40具有RBCC泛素连接酶结构域,作者推测TRIM40直接靶向ROCK1进行降解。
值得注意的是, MG132蛋白酶体抑制剂阻止了TRIM40介导的ROCK1蛋白水平降低,而溶酶体抑制剂氯喹没有效果(图3D)。此外,在MG132处理后,表达TRIM40的细胞显示出TRIM40与ROCK1的相互作用以及ROCK1泛素偶联物的大量积累(图3E,F)。
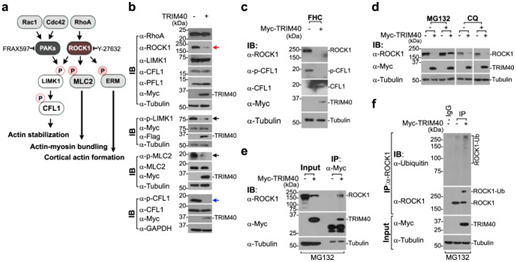
图3:TRIM40通过促进ROCK1降解来调节RhoA–ROCK1信号转导。
(A) Rho家族GTP酶的肌动蛋白细胞骨架信号通路示意图。(B) 免疫印迹实验检测表达对照载体或Myc-TRIM40的HT-29细胞中与肌动蛋白细胞骨架相关的信号蛋白的表达。(C) 免疫印迹分析检测表达对照载体或Myc-TRIM40的FHC细胞中ROCK1、p-CFL1和CFL1的蛋白水平。(D) 用MG132(29μM)或氯喹(CQ,40μM)处理1小时后,免疫印迹分析检测表达对照载体或Myc-TRIM40的HT-29细胞中ROCK1的蛋白水平。(E) 在表达对照载体或Myc-TRIM40的HT-29细胞中对TRIM40和ROCK1进行免疫共沉淀。(F) 免疫印迹实验检测表达对照载体或Myc-TRIM40的HT-29细胞中ROCK1泛素化水平。
04
TRIM40 的RBCC结构域是 ROCK1 降解所必需的。
为了进一步确定TRIM40 哪个功能结构域负责降解ROCK1,作者构建了TRIM40ΔRING、TRIM40ΔBB、TRIM40ΔCC或TRIM40ΔCT的 TRIM40 突变体(图4A)。TRIM40ΔRING未能诱导ROCK1泛素化(图4B),导致ROCK1蛋白水平部分恢复(图4C)。通过E3连接酶死亡的TRIM40突变体(TRIM40-C29S)也间接证明TRIM40ΔRING失去功能活性,导致TRIM40未能降解ROCK1(图4D)。共聚焦分析还显示,在表达TRIM40-C29S的细胞中,皮质肌动蛋白也出现了类似的部分恢复,正如在TRIM40ΔRING的细胞中观察到的那样(图4E)。这一发现表明,TRIM40的E3连接酶活性对于引导ROCK1通过泛素-蛋白酶体途径进行降解是必要的。此外,TRIM40ΔBB完全失去了对ROCK1的结合亲和力(图4F),导致TRIM40未能驱动ROCK1降解和皮质肌动蛋白破坏(图4C,E)。TRIM40ΔCT的二聚体形式的产生使作者推测C末端区域在结构上阻止了TRIM40二聚化(图4C,F),这可能是影响其破坏上皮完整性能力的决定性因素。事实上,TRIM40ΔCT对ROCK1降解或相互作用没有影响(图4C,F),并最终失去了其皮质肌动蛋白破坏功能(图4E)。此外,TRIM40ΔBB和TRIM40ΔCT均未使细胞间距离缩小,并且TRIM40ΔRING仅部分影响了细胞间的距离(图4G),表明TRIM40突变体影响ROCK1降解的功能能力直接反映在皮质肌动蛋白形成和细胞间距离中。综上所述,这些结果表明,TRIM40通过RBCC结构域与ROCK1相互作用,作为E3连接酶直接靶向ROCK1,并且促进ROCK1的降解从而导致上皮细胞完整性的破坏。
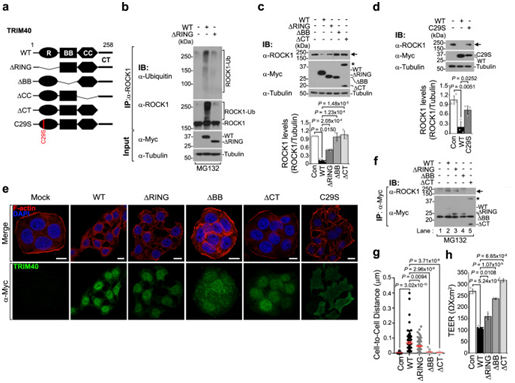
图4:TRIM40的RING结构域、B box结构域和C端结构域是降解ROCK1所必需的结构。
(A) 全长TRIM40(WT)和TRIM40突变体的结构示意图。(B) 免疫印迹显示表达对照载体、WT-TRIM40或TRIM40∆RING细胞中ROCK1的泛素化水平。(C) 免疫印迹实验检测表达WT-TRIM40或TRIM40突变体的HT-29细胞中ROCK1的蛋白水平。(D) 免疫印迹检测表达对照载体WT-TRIM40或TRIM40-C29S的HT-29细胞中ROCK1的蛋白水平。(E) 共聚焦图像显示表达对照载体、WT-TRIM40、TRIM40缺失或TRIM40-C29S的HT-29细胞中皮质F-肌动蛋白的水平。(F) 在表达对照载体、WT-TRIM40或TRIM40缺失突变体的HT-29细胞中对TRIM40突变体和ROCK1进行免疫共沉淀。(G) 关于表达对照载体、WT-TRIM40、TRIM40∆RING、TRIM40∆BB box和TRIM40∆CT的HT-29细胞间距离的统计分析。(H) TEER实验检测表达对照载体WT-TRIM40或TRIM40缺失突变体的Caco-2细胞的肠上皮屏障功能。
05
Trim40−/−小鼠不易发生DSS诱导的结肠炎。
研究结果表明,经DSS诱导的小鼠结肠组织Trim40表达显著增加(5A)。为了进一步证实这一点,作者通过特异性识别小鼠Trim40 mRNA的RNA探针进行RNA原位杂交测定,观察到DSS处理的小鼠直肠和结肠组织中Trim40表达显著增加(图5B)。作者构建了Trim40−/−小鼠,在DSS诱导之后,Trim40−/−小鼠出现UC的临床症状,如体重减轻和结肠长度缩短,但这些症状并没有对照小鼠严重(图5D)。尽管Trim40 −/−小鼠体重有所下降,但在DSS去除后逐渐恢复,并未在对照小鼠中观察到类似情况(图5C)。由于UC症状严重,大多数对照小鼠在DSS给药后10天内死亡,而约70%的Trim40−/−小鼠存活期超过2周(图5E)。组织病理学还证实,在DSS诱导后,Trim40−/−的小鼠肠道上皮屏障、隐窝和杯状细胞结构保持完整,固有层或上皮中未观察到可见的淋巴细胞浸润(图5F);相比之下,对照小鼠表现出严重的上皮屏障破坏,杯状细胞较少,隐窝变形,在直肠和远端结肠中都清楚地观察到淋巴细胞的广泛浸润(图5F);此外,DSS能够诱导小鼠体内的促炎细胞因子和趋化因子表达,与对照组相比,Trim40−/−小鼠产生的细胞因子显著减低,除外抗炎细胞因子IL-10(图5G)。与人上皮细胞的体外结果一致,DSS处理的对照小鼠由于TRIM40上调ROCK1(mROCK1)蛋白水平显著降低;然而,无论DSS给药如何,在Trim40−/−小鼠中未观察到mROCK1表达水平的降低(图5H)。总的来说,体外和体内结果表明,TRIM40促进ROCK1降解和随后的F-肌动蛋白不稳定,导致肠上皮屏障功能的丧失。
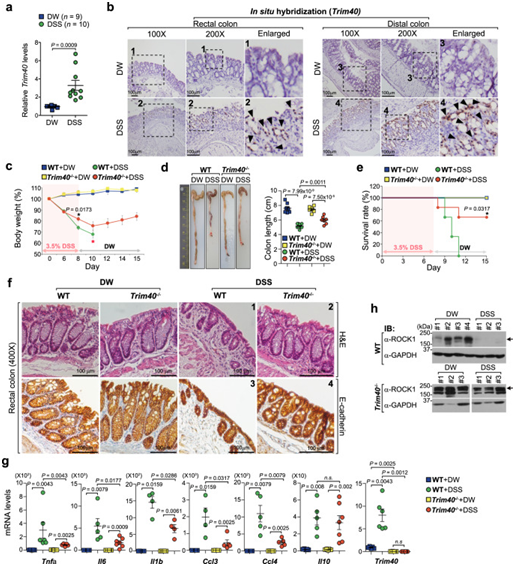
图5:缺乏Trim40的小鼠不易发生DSS诱导的结肠炎。
(A) qPCR检测2% DSS 诱导的结肠炎中小鼠结肠组织Trim40的mRNA水平。(B) ISH显示2% DSS给药后的WT 雄性小鼠直肠或远端结肠组织中的Trim40 mRNA水平。(C) 在3.5% DSS给药后检测WT和Trim40−/−的雄性小鼠体重减轻情况。(D) 雄性小鼠在2%DSS给药后第7天的结肠长度图像。(E) 用3.5% DSS处理8天,然后用正常水处理7天的雄性小鼠的存活曲线。(F) 在3% DSS后第1天对WT和Trim40−/−的雄性小鼠的直肠E-钙粘蛋白进行H&E染色和IHC染色。(G) qPCR检测2%DSS诱导的结肠炎中小鼠结肠组织相关基因的mRNA水平。(H) 免疫印迹实验检测2.5%DSS持续3天给药后的WT和Trim40−/−的雄性小鼠直肠区域的ROCK1蛋白水平。
+ + + + + + + + + + +
结 论
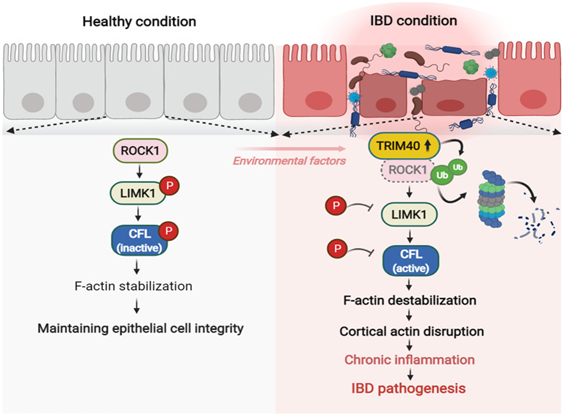
1、健康状态下,TRIM40并不表达从而维持ROCK1的正常表达,ROCK1磷酸化LIMK1,从而使CFL失活,进而维持肠道上皮的完整性。
2、IBD发生时,表观遗传沉默的 TRIM40的表达上调,直接靶向ROCK1降解和皮质肌动蛋白破坏,加速破坏上皮完整性和功能,最终导致严重的肠道组织损伤。
3、TRIM40可能代表一种潜在的生物标志物,用于使用直肠组织活检诊断UC和CD以及限制IBD发生和发展的治疗靶点。
+ + + + +



