

 English
English文献解读|Cell Rep(8.8):甲状腺未分化癌的基因组和进化景观
✦ +
+
论文ID
原名:The genomic and evolutionary landscapes of anaplastic thyroid carcinoma
译名:甲状腺未分化癌的基因组和进化景观
期刊:Cell Reports
影响因子:8.8
发表时间:2024.02.26
DOI号:10.1016/j.celrep.2024.113826
背 景
人类恶性肿瘤的致死率差异很大。一些肿瘤类型,如分化型甲状腺癌 (DTC) 和前列腺腺癌,通常是惰性的,具有较长的生命史,可以通过微创治疗甚至监测方案进行治疗。甲状腺未分化癌(ATC)是最致命的人类恶性肿瘤之一,它通常与分化型甲状腺癌同时发生,ATC具有极大的侵袭性:约80%的ATC发生在既往甲状腺癌病史的背景下,或与DTC具有独特共同发生区域(co-DTC),最常见的是乳头状甲状腺癌(PTC),但其侵袭性的分子起源尚不清楚。
实验设计
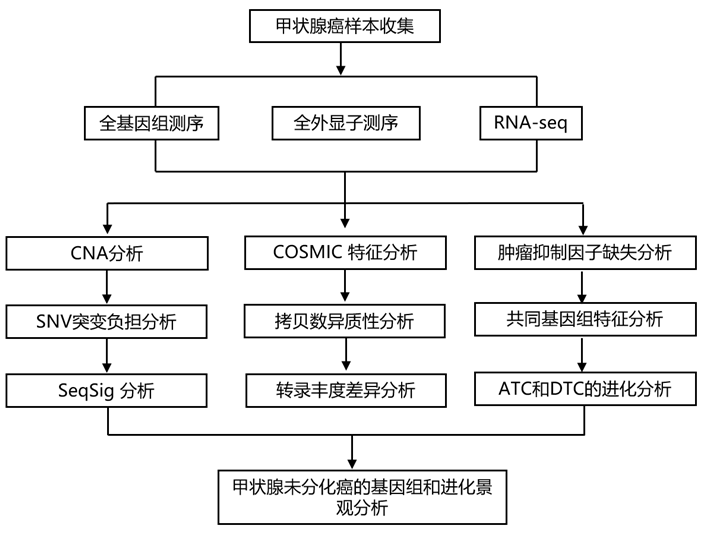
结 果
01

ATC 表现出中等的 SNV 突变负担
研究团队收集了 292 名患者的 329 个甲状腺癌样本,其中 213 个来自 ATC 患者,115 个来自 PTC 患者。通过 SNP 微阵列对样本进行了拷贝数畸变 (CNA) 特征分析,并通过全外显子组 (WES) 和全基因组测序 (WGS) 以及 RNA 测序 (RNA-seq) 来鉴定单核苷酸变异 (SNV)。在这个庞大的全基因组ATC队列中,他们鉴定出3.8±1.2 SNV/Mb的DNA序列(图1A-C),伴有120±44个CNA(图1B-D)。平均CNA为6.21±1.33 Mb。相对于32种肿瘤类型,27种ATC比ptc显示更多的CNA(图1D)和更多的snv /Mb(图1C),但比大多数其他成人癌症类型少。尽管它们具有临床侵袭性,但它们并不是最高度突变的肿瘤类型之一,也没有表现出相对于其他癌症类型的非典型的高肿瘤间变异性。
SeqSig FDR最小的前5个基因分别是TP53、NRAS、BRAF、PIK3CA和USH2A(图1A)。原发性ATC中TP53的非功能性或部分功能性非同义snv与mRNA丰度升高相关。
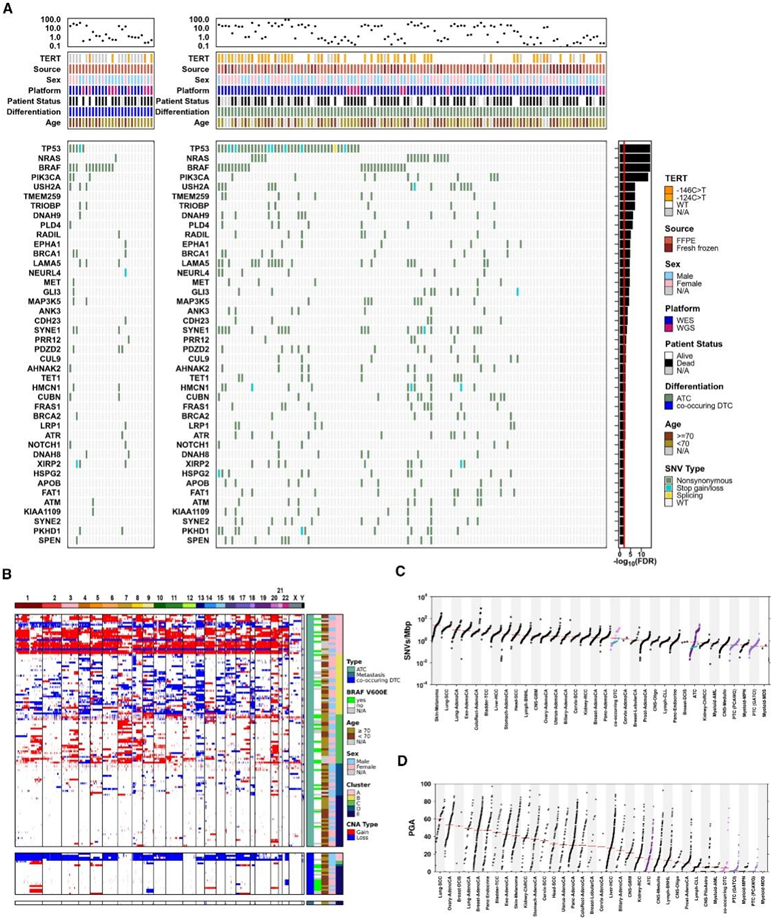
图1. 甲状腺未分化癌的体细胞突变情况。
(A) 甲状腺未分化癌 (ATC) 的突变密度(每百万个 DNA 碱基对的单核苷酸变异 [SNV])差异很大。(B) ATC 中的拷贝数畸变 (CNA);共识聚类用于确定样本分组的最佳方法和名称。(C-D) 将 ATC、同时发生的 DTC 和乳头状甲状腺癌 (PTC) 的突变密度指标与 PCAWG(全基因组泛癌症分析)数据集中提供的 32 种其他肿瘤类型进行比较:SNV /Mb 和 总 PGA。
02
ATC 中的拷贝数异质性和转录组学特征
单个ATC在其潜在的突变过程中差异很大,具有激活的COSMIC特征1、5、6和13以及罕见的新特征(图2A)。最后一种以G[T>G]G突变为特征,仅在三种肿瘤中检测到(图2B)。据报道,其他类型的甲状腺癌具有COSMIC特征1、5和13的激活,而ATC反映了这些特征。COSMIC 5描述了由低频嘧啶过渡突变组成的多种点突变,COSMIC 13归因于AID/APOBEC活性,但令人惊讶的是,这些特征都与性别、年龄、CNA亚型、TERT启动子状态或总生存期无关。因此,ATC比其他甲状腺癌有更多的体细胞snv,但比其他类型的癌症少,没有广泛复发的驱动突变或突变过程。
相比之下,ATC具有多重复发性染色体规模事件的背景,包括先前报道的13号染色体丢失和20q号染色体获得。20q上的一个基因亚群的过表达可促进甲状腺癌的进展。他们还发现了一种新的7号染色体的复发性增益。共识聚类显示,这些大规模变化反映了5种不同的CNA亚型,分别为A-E(图1B,图2C)。每种亚型都是由特征性的基因组异常来定义的,例如亚型A的大规模扩增和偶尔的臂水平缺失(11%的ATC)以及单株多染色体的大规模缺失,类似于甲状腺嗜瘤性癌(OCT),为B亚型。亚型C(20%)以7号和20号染色体的增益(类似于OCT)和1q增益(类似于PTC25)为主,而亚型D(15.5%)表现出许多CNA的病灶区域。E亚型中拷贝改变的缺失并非由于广泛的间质浸润,病理检查时估计的肿瘤纯度进一步支持了这一点。在11例集中病理复查的E亚型ATC肿瘤中,评估肿瘤纯度中位数为50。因此,与乳腺癌、前列腺癌和头颈癌的相似特征类似,E亚型显示出静止的CNA结构(图2C)。
这些亚型与总体基因组不稳定性(图1D)、年龄、TP53突变状态、肿瘤纯度和倍性相关。这些广泛的变化伴随着许多高度复发的病灶驱动CNA(图2E)。
接下来,他们结合来自 TCGA 的正常、原发性 PTC 和转移性 PTC RNA-seq 数据评估了 ATC和细胞系之间的转录丰度差异。他们观察到正常甲状腺、PTC 和 ATC 的甲状腺分化评分(TDS)逐渐下降,表明进行性去分化(图 2 G)。他们还在本项研究的RNA-seq数据集中评估了BRAF-RAS评分。尽管TCGA内的原发性ptc在BRAF和ras改变的肿瘤之间表现出明显的分离,但ras突变的ATC肿瘤表现得更像BRAF,与先前的研究相似(图2H)。
他们使用MCPCounter进一步估计了样品中基质细胞和免疫细胞群的丰度。与正常甲状腺相比,原发性ptc巨噬细胞/单核细胞和T细胞丰度降低,中性粒细胞丰度增加(图2I)。另一方面,ATC的肿瘤微环境与原发性和转移性PTC不同,其成纤维细胞和巨噬细胞/单核细胞的丰度显著升高,内皮细胞、骨髓树突状细胞和中性粒细胞的丰度显著降低(图2I)。

图2. ATC的基因组特征及其与临床特征的关联。
(A) 非负矩阵分解 (NMF) 识别出 ATC 内的五个三核苷酸特征,其中四个与已知的 COSMIC 特征相匹配,一个是新特征。(B) 对于每个患者,贡献每个特征的 SNV 的比例。(C-D) 每个 ATC 亚型、同时发生的 DTC 和 PTC 样本的平均 CNA 特征和 PGA 分布。(E) GISTIC 用于识别 ATC 内经常出现的 CNA。(F) 检验来评估 ATC 中基因组特征的重叠。(G–I) ATC和细胞系的 RNA-seq 样本之间甲状腺分化评分、BRAF-RAS 评分和估计免疫细胞亚群 (I) 的比较。
03
综合基因组学分析表明缺乏 SNV 驱动突变的 ATC 中肿瘤抑制因子的频繁缺失
为了确定 ATC 发病机制中同时发生的突变过程,他们分析了突变密度、突变特征和驱动基因特征的相互关联(图 2 F)。检测到多种关联,包括 SNV 突变密度(SNV/Mb序列)与 COSMIC 特征1 和 5之间的强相关性。
他们对ATC/co-DTC肿瘤子集进行了综合分析,在之前的甲状腺癌研究或SeqSig复发基因中强调的通路(如MAPK-RAS、SWI/SNF、肿瘤抑制因子等)中未发现SNV改变(总共21个ATC和5个co-DTC肿瘤)(图3)。该基因集合中缺乏SNV改变不是由于肿瘤纯度或测序覆盖率的差异。对CNA状态的分析显示,10例ATC肿瘤存在肿瘤抑制因子CDKNA2、RB1、CDK7或BRCA2中的一种缺失(通常是多重改变)(图3)。这些结果表明这些肿瘤抑制因子的拷贝数缺失可能是疾病进展和去分化的潜在驱动因素。
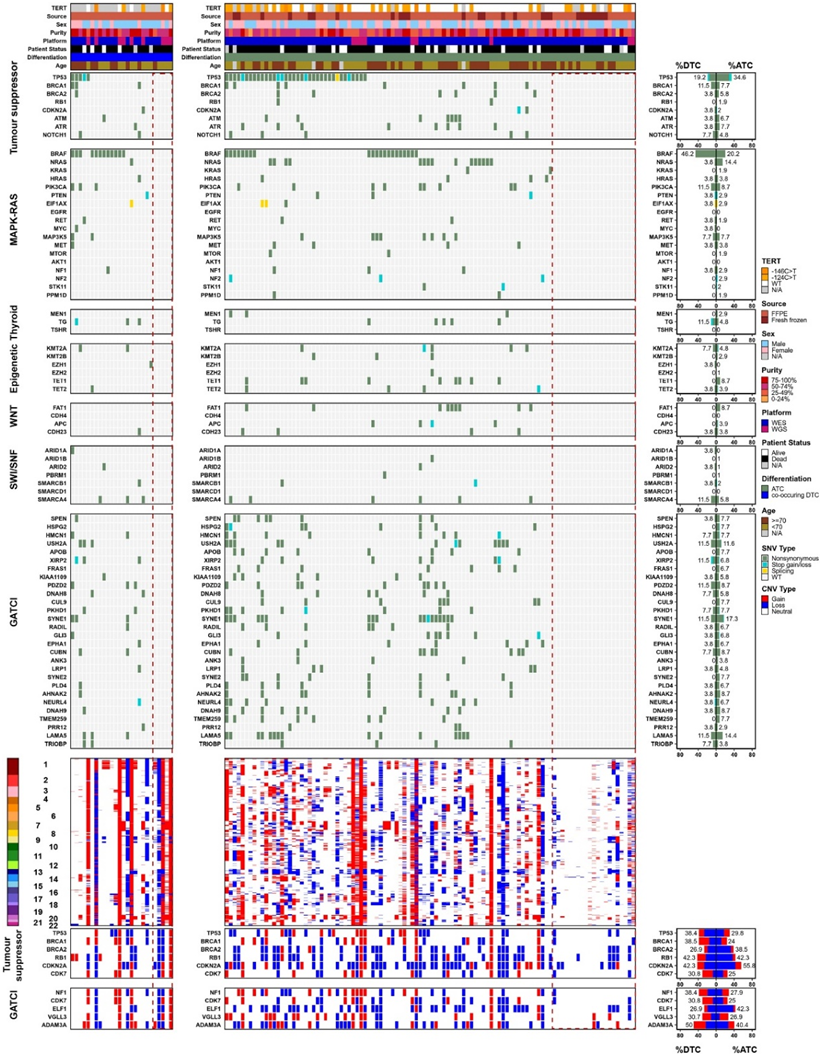
图3. 未分化甲状腺癌的基因组特征涉及多种途径。
04
ATC 和 DTC 具有共同的基因组特征
他们使用本研究中新分析的 PTC 样本和 TCGA 数据集比较了 PTC、co-DTC 和 ATC 之间的 SNV 和 CNA。PTC的两个复发臂级事件是1q扩增和22q缺失,这两种情况在 ATC 中以及 ATC 中同时出现的 DTC 组件中出现的频率相似(图 1 B ,图4 C)。所有PTC的复发性局灶性CNA也在ATC中检测到,但几种CNA驱动因素在ATC中明显更频繁(图4A)。CDKN2A的缺失在共同发生的dtc和ATC中都是复发性的,但在ptc中很少见(图4B)。BRCA2在ATC中也经常缺失(33.6%),在dtc中不常见(13.6%),在ptc中罕见(4.5%),RB1也是如此,BRCA2和RB1位于同一染色体臂上。其他几个区域在ATC和共同发生的dtc中显示出更高的拷贝数变化率,包括染色体20q上的一个广泛区域,其中包含328个基因,相对于两组ptc, ATC中优先扩增。
接下来,他们比较了与ATCs同时发生的dtc的体细胞CNA和SNV谱(图4C-D)。与ATC共同发生的dtc显示,与分离的dtc相比,其基因组受CNA影响的比例更大。同样,与未诊断为ATC的个体中分离的ptc或其他甲状腺癌相比,共同发生的dtc含有更多的snv:它们的总体突变密度在统计学上与ATC无法区分。为了探索体细胞驱动snv,他们将当前的研究与之前的六项ATC、PTC和OCT的测序研究结合起来。BRAF是分化型甲状腺癌中唯一更常见的基因:它在50.9%的ptc、50.0%的合并dtc和21.3%的ATC中发生突变(图4E)。事实上,在匹配的共发生病例中,所有BRAFV600E变体的一半仅在DTC成分中检测到,而不在ATC样本中检测到。相比之下,多个其他驱动因素在ATC和共同发生的dtc中优先发生突变,包括ATM、ATR、BRCA2、PIK3CA和TP53(图4E)。例如,TP53突变频率从ptc的0.9%到合并dtc的21.4%和ATC的36.8%不等。BRAFV600E突变在PTC中(相对于ATC)显示出更高的频率,这表明这种突变更早发生,并且更倾向于克隆性进化,而NRAS突变在两者(PTC和ATC)之间没有差异。

图4. ATC 和DTC 具有相同的克隆起源。
(A) GISTIC 用于识别 PTC 内重复出现的 CNA;与 ATC 相比,这表明 17 号染色体的多次扩增是显著且经常发生的。(B) GISTIC 用于识别同时发生的 DTC 中复发的 CNA。(C) 21 名患者具有通过多个肿瘤区域的拷贝数阵列生成的 CNA 图谱。(D) 30 名患者在多个地区进行了 WES 或 WGS。(E) 比较数据集(GATCI-ATC、GATCI-DTC、GATCI-PTC 和 TCGA-PTC)之间的基因突变频率,以确定肿瘤进展的候选驱动因素。(F) 通过全基因组测序确定患有 ATC 和配对同时发生的 DTC 的患者的 CNA 景观。
05
ATC 和 DTC 在同一个致突变环境中平行进化
因此,他们利用配对的ATC和共同发生的DTC多区域WGS数据,通过验证的snv亚克隆重建管道和亚克隆CNA状态,推断每个患者的克隆层次和突变时间(图4D)。WES数据显示,编码SNV变异的等位基因频率上显示出高度的相关性。
在WGS数据中,发现每个肿瘤都表现出独特的进化史,但每个病例都有两个共同特征。co-dtc和ATC具有共同的克隆起源,共同的祖先分裂形成不同的谱系,具有显著的亚克隆多样化(图5A-J)。这种ATC和DTC谱系的分裂通常出现在肿瘤进化的早期:共同祖先拥有约95%的CNA,但只有19.1%±7.9%的snv。DTC区域并不局限于特定的CNA亚型,并且在PGA、CNA总数或其他特征上没有一致的趋势(图4F),这与缺乏克隆相互作用相一致。
在大多数情况下,空间分离的ATC成分不包含DTC克隆,如ATCWGS37(图5B)和ATCWGS34(图5C),在本项研究的检测极限下,每种情况下快速生长的ATC只显示一个亚克隆。在一些情况下,他们能够清楚地识别出一个致突变位置,其中大部分细胞具有相似的致癌改变,例如ATCWGS34(图5C)和ATCWGS33(图5D)。在这些病例中,空间上不同的肿瘤在ATC和共同发生的DTC中都有共同的突变。ATCWGS33也为两种肿瘤成分之间亚克隆相互作用的复杂性提供了一个显著的例子。肿瘤从一个共同的肿瘤位置分化成两个分支(亚克隆2和亚克隆3)(图5D)。分化肿瘤区和间变性肿瘤区都包含来自这些肿瘤分支的克隆,但具有显著的差异进化:比较亚克隆3(在DTC中发现的频率较低)和亚克隆6(在ATC中发现的频率较高)。
亚克隆重建分析表明,ATC在主要 DNA CNA 积累之后从 DTC 亚克隆进化而来(图 5 K)。然后,该克隆获得了特有的额外致癌驱动因素及其大部分体细胞改变。原始DTC克隆和诱变领域的其他亚克隆可以作为克隆混合物继续在ATC中共存,但只有ATC克隆具有转移潜力。
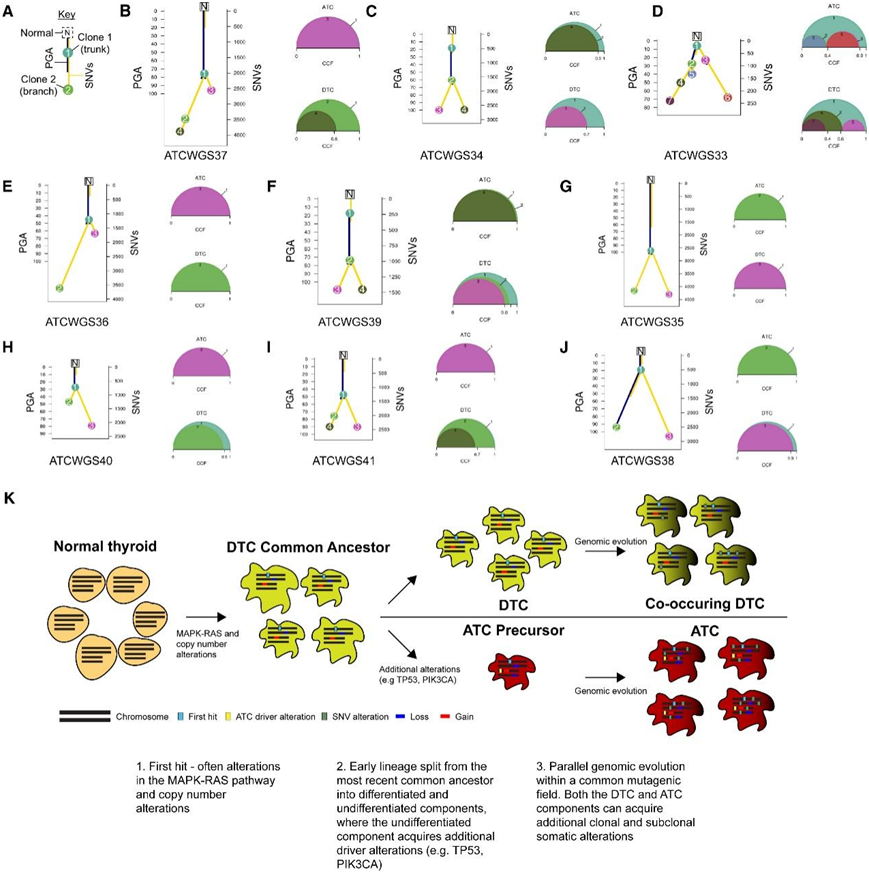
图5. ATC和DTC的进化分析。
(A) 解释图的关键。(B–J) 使用全基因组测序对 9 名患者进行亚克隆重建。(K) 提出的甲状腺癌进展模型。
+ + + + + + + + + + +
结 论
本项研究对甲状腺癌 329 个区域的肿瘤 DNA 进行了测序,其中 213 个区域来自原发性未变性甲状腺癌患者。他们还使用分化型甲状腺癌和未分化型甲状腺癌成分的多区域测序对 9 名患者进行了全基因组测序。利用这些数据,他们证明未分化甲状腺癌比其他甲状腺癌具有更高的突变负担,具有独特的突变特征和分子亚型。此外,不同的癌症驱动基因在未分化和分化的甲状腺癌中发生突变,甚至是在单个患者中出现的癌症。最后,明确证明,未变性甲状腺癌与同时发生的分化癌具有相同的基因组起源。
+ + + + +



