

 English
English文献解读|(33.883):肾上腺素B2受体酪氨酸激酶是原癌基因MYC和巴雷特肿瘤核心分子程序的调节剂
✦ +
+
论文ID
原名:The Ephrin B2 Receptor Tyrosine Kinase Is a Regulator of Proto-oncogene MYC and Molecular Programs Central to Barrett’s Neoplasia
译名:肾上腺素B2受体酪氨酸激酶是原癌基因MYC和巴雷特肿瘤核心分子程序的调节剂
期刊:Gastroenterology
影响因子:33.883
发表时间:2022.11
DOI号:10.1053/j.gastro.2022.07.045
背 景
食管腺癌(EAC)是一种侵袭性恶性肿瘤,是全世界癌症相关死亡的主要原因之一。其5年生存率很低,而且绝大多数EAC患者确诊时到终末期,已失去手术机会。由于缺乏EAC发生发展相关的知识,目前仍无有效的靶向治疗。EAC通常起源于Barrett食管(BE),这是一种癌前病变,为远端食管的鳞状上皮(SQ)被肠型柱状化生所取代导致。尽管已经提出了几种关于产生BE的起源细胞的模型,但BE-EAC发展表型的分子因素仍然未知。因此,研究BE-EAC发生发展的机制将有助于开发有效的预后生物标志物,以及早期化学预防/治疗策略。在这里,作者使用一种独特的系统生物学方法,结合癌前病变和EAC活检组织中获得的基因组转录组谱,试图在BE病变早期的通路水平上识别最频繁的调控网络,进行了分子和表型研究,以剖析相关非恶性、癌前和恶性哺乳动物模型系统中选择的网络成分的功能意义。
实验设计
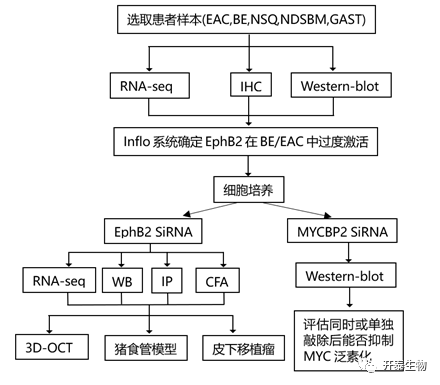
结 果
01

EphB2信号子网在几乎所有BE和EAC中都过度激活
首先,作者使用InFlo系统生物学框架,根据来自预处理EAC/BE和非恶性(正常SQ和胃)活检组织中得到的RNA-seq数据,确定了BE-EAC发展中最常发生变化的通路网络。发现EphB2酪氨酸激酶信号在100%的BE和88%的EAC中过度激活(图1A),与正常SQ和胃组织相比,BE/EAC病变中选择性地诱导EphB2受体,同时EphB配体(EFNB1和EFNB2)略有增加(图1B)。
为了测试EphB2在BE和EAC中诱导的共性,他们对单纯非恶性、癌前病变和EAC活检组织(n=885)进行RNA-seq分析发现,与正常食道SQ和胃组织相比,BE化生/异型增生/腺癌组织中EphB2的诱导显著(>8倍)并有差异(P<0.0005) (图1C)。IHC分析亦证实在BE和EAC病变中选择性地诱导了EphB2蛋白的表达(图1D)。免疫印迹分析显示,BE-EAC细胞中EphB2蛋白的表达高于正常食道SQ细胞系,而且EFNB1/B2配体在SQ、BE和EAC中都有表达,尤其在EAC细胞中表达水平较高(图1E)。
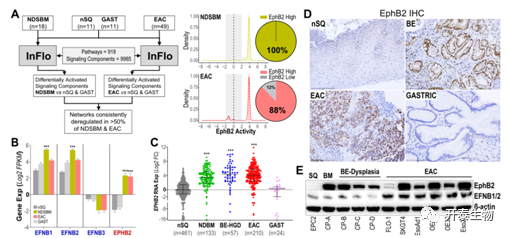
图1. BE和EACs中EphB2信号的过度激活
02
EphB2对原癌基因MYC的调控
接下来,为了了解EphB2信号下游的介质和效应通路,作者对EphB2进行基因敲除并对具有代表性的EAC和BE细胞系进行了RNA-seq和InFlo分析。发现除了先前在BE/EAC中涉及的通路, c-MYC活性和相关的转录网络受EphB2正向和显著调节(图2A)。有趣的是,EphB2不调节MYC RNA的表达(图2B)。在EphB2基因敲除后,通过对EAC(SKGT4,OE33)和BE模型(CP-A,CP-D)进行qPCR和WB分析进一步评估,发现SKGT4和CP-D在EphB2基因敲除后MYC RNA表达无变化,而OE33和CP-A则相对下降(图2C)。然而,WB分析显示,在所有BE和EAC细胞系中,EphB2基因敲除后MYC蛋白明显受到抑制(图2C),而EphB2的诱导与原发BE/EAC病变中MYC蛋白的阳性相关(图2D)。值得注意的是,先前的研究结果进一步表明,EphB2对MYC蛋白本身具有直接调节作用,是调节的主要模式之一。具有代表性的EAC(SKGT4)细胞系中EphB2的敲除加速了MYC的降解(图2E),进一步支持了EphB2对MYC的翻译后调控。
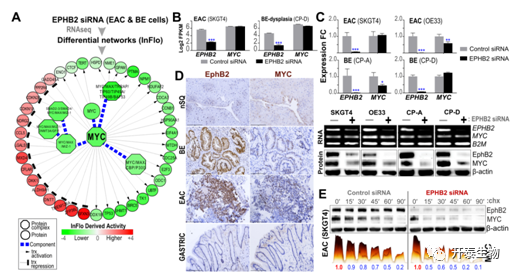
图2. MYC是EphB2信号的下游靶点
03
EphB2与MYC结合蛋白2相互作用,并通过泛素-蛋白酶体途径调节MYC
为了进一步探讨EphB2对MYC蛋白调控的机制,作者在一个具有代表性的EAC系(SKGT4)中,用原始EphB2抗体或对照IgG抗体以及EphB2稳定重组和不稳定重组进行了IP。通过质谱分析确定MYC结合蛋白2(MYCBP2)是内源性EphB2结合蛋白的结合伙伴(图3A),并用正反向IP-WB分析验证了这一点(图3B)。
MYCBP2是一种非典型的E3泛素蛋白连接酶,它介导靶蛋白上苏氨酸/丝氨酸残基的泛素化。值得注意的是,MYCBP2与MYC的反式激活结构域相互作用,被推测为促进或以其他方式调节MYC的活性。EAC细胞中的CO-IP研究确实证实了内源性MYCBP2-MYC相互作用(图3C)。此外,与对EphB2的发现相反(图2B和C),MYCBP基因的敲除与EAC、BE化生和异型增生细胞系中MYC蛋白的增加有关(图3D),表明它在Barrett瘤中起到MYC负调控的作用。相反,MYC的敲除并不影响EphB2或MYCBP2,表明EphB2和MYCBP2在调节级联中都位于MYC的上游(图3E)。
为了进一步了解EphB2、MYCBP2和MYC之间的功能关系,他们在EAC/BE细胞中进行了EphB2和MYCBP2的敲除。值得注意的是,MYCBP2基因敲除与EphB2沉默同时发生,并不能完全恢复仅在MYCBP2基因敲除中观察到的MYC水平(图3F)。鉴于MYCBP2是一种E3泛素连接酶,沉默EphB2或MYCBP分别导致MYC泛素化增加或几乎全部抑制(图3G)。与单独敲除MYCBP2相比,与EphB2沉默同时丢失MYCBP2并不能完全抑制MYC泛素化(图3G)。总之,这些发现强烈表明,EphB2在翻译后调节MYC的稳定性/活性,部分是通过它与MYCBP2和泛素-蛋白酶体途径的相互作用。
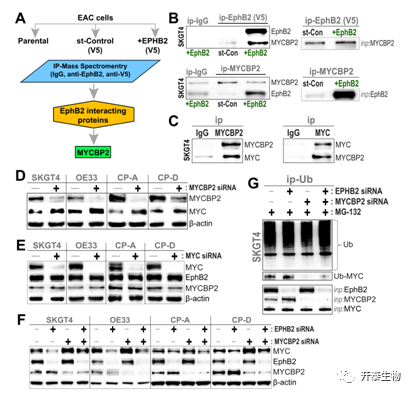
图3. EphB2与MYCBP2相互作用
04
EphB2信号影响EAC和BE细胞的生长/活性
接下来,作者通过在不同的EAC细胞模型中进行EphB2基因敲除来评估EphB2信号的表型后果,发现EphB2基因敲除显著阻碍了多种EAC细胞的增殖能力(图4A)。为了进一步证实该结果,他们用稳定表达Dox诱导的EphB2 shRNA的EAC细胞(SKGT4)重复了该分析,并再次发现EphB2基因的敲除显著减少了EAC的菌落生长(图4B)。此外,EphB2基因敲除显著降低了MYC蛋白水平并阻碍了体内EAC肿瘤移植瘤的生长(图4B)。
为了明确EphB2相关的生长依赖是否特定于恶性阶段,或者它们在先前的BE病变中是否明显,作者在BE化生(CP-A)和BE异型增生(CP-D)细胞中进行了EphB2 siRNA敲除。发现结果与先前观察类似,EphB2的敲除显著降低了BE-化生和-异型增生细胞的存活率(尽管程度较小)(图4C)。
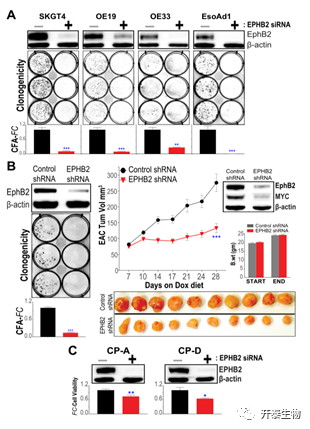
图4. EphB2影响EAC和BE细胞的生长特性
05
EphB2激活干扰正常食道SQ上皮成熟并促进BE相关分子程序
hTERT永生化食管SQ细胞(EPC2)在作为食管3D器官型培养(OTC)生长时形成分层鳞状细胞层,模拟体内食管形态和稳态。永生化的EPC2细胞用EphB2或填充物对照载体稳定重组,尽管对照EPC2细胞如预期的那样分化并成熟为持久的复层SQ上皮,即使对细胞增殖没有影响,但EphB2的重组扰乱了SQ-OTCs的形态和正常成熟(图5A)。对BE相关柱状分化标记的评估显示,在EphB2重组的EPC2 OTC和单层培养中SOX9和FOXA2的表达增加(图5A和B),而FOXA2在EPC2-SQ和BE细胞中均受到EphB2的一致调节(图5B)。EphB2的激活也导致了p63的适度下降(图5B),这是一种对鳞状上皮的发育和动态平衡至关重要的转录因子。重要的是,EPC2-SQ细胞中EphB2的重组增强了OTC和单层培养中的MYC蛋白(图5A和B),同样对MYC RNA水平没有显著影响(图5C)。与此一致,EphB2也适度影响MYC的降解(图5C),并显示出与SQ细胞中MYCBP2的相互作用(图5D)。
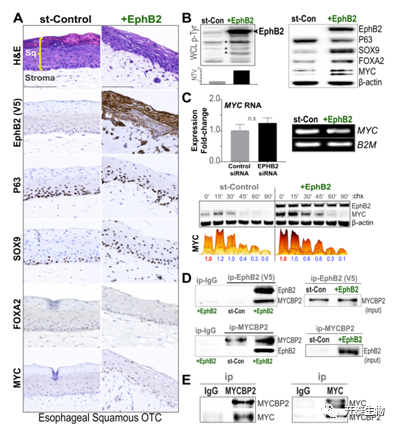
图5. EphB2的激活破坏了食道SQ上皮的成熟
06
EphB2存在于EAC相关的食道粘膜下腺中,这是BE前体的另一个潜在来源
鉴于上述观察,接下来作者评估了EphB2和MYC在食道粘膜下腺(ESMGs)中的作用,ESMGs是一组参与食管损伤修复的前体细胞。因此,他们首先评估了ESMGs中EphB2的状态(这些ESMGs是从EAC患者接受化疗前手术切除的食管组织中获得的),在与EAC相关的ESMG中观察到了EphB2和MYC选择性的稳健模式(图6A)。接下来他们将自手术切除的单纯非EAC食道组织作为额外的对照,结果显示EphB2和MYC缺失。除此之外,他们使用源自猪食管的ESMGs的配套球体培养模型,进行单细胞RNA-seq和Inflo分析来评估EphB2-MYC的活性,发现BE样球体的EphB2和MYC活性显著高于SQ样ESMG球体,与细胞周期时相无关(图6B)。这些发现进一步表明,在前体细胞中诱导EphB2-MYC活性可能是BE发育的关键步骤。
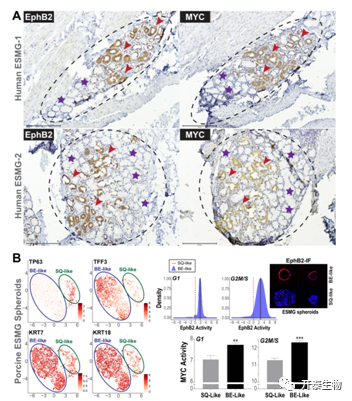
图6. EphB2-MYC在ESMG相关ADM和猪类ESMG球体中的激活
07
MEK1小分子抑制剂在体内抑制MYC活性和EAC肿瘤的生长
上述研究结果表明EphB2-MYC轴是EACS潜在的脆弱治疗靶点,然而,尚无临床批准的针对EphB2或c-MYC的抑制剂。有效的临床批准的MEK1抑制剂(Meki) 考比替尼在EAC细胞系中显示出显著的细胞毒性,因此作者在多种肿瘤异种移植模型中评估了考比替尼的抗肿瘤效果,发现其可导致体内EAC肿瘤生长的强烈抑制和/或消退,而且肿瘤中MYC蛋白显著减少(图7)。这些发现表明,对Meki的反应阈值可能取决于ERK、MYC和/或其他下游效应器的相对/联合水平,靶向MYC相关的上游信号轴可能是一种有益的EAC新治疗策略。
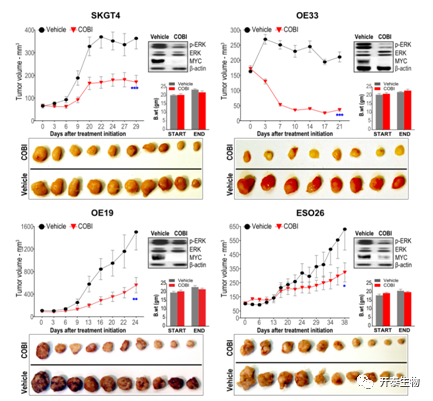
图7. 抑制MEK可抑制MYC活性和体内EAC肿瘤生长
+ + + + + + + + + + +
结 论
总之,该研究确定EphB2信号在BE-EAC连续体中经常过度激活,EphB2是BE-EAC病理生物学的潜在关键决定因素,其作为MYC的上游调节因子,EphB2-MYC轴的激活可能先于BE的发展。靶向EphB2 / MYC可能是这种通常难治性和侵袭性癌症的一种有前途的治疗策略。
+ + + + +



