

 English
English文献解读|Cell(42.5):免疫-微生物组协调决定了健康人体内干扰素的设定点
✦ +
+
论文ID
原名:Immune-microbiome coordination defines interferon setpoints in healthy humans
译名:免疫-微生物组协调决定了健康人体内干扰素的设定点
期刊:Cell
影响因子:42.5
发表时间:2026.03.09
DOI号:10.1016/j.cell.2026.02.003
背 景
个体间免疫差异对人类健康和疾病具有广泛的影响。人群研究已开始识别免疫变异的驱动因素,结果表明环境因素的影响可能超过遗传因素。更深入的理解有助于揭示疾病易感性、严重程度以及对免疫调节疗法(包括疫苗接种、癌症免疫疗法和自身免疫性疾病的免疫抑制治疗)的不同反应。肠道微生物群对免疫系统的形成起着至关重要的作用;然而,其与健康人群基础免疫状态的关系仍未完全阐明。
实验设计
结 果
01

健康人群中免疫和微生物组的变异
为了研究人类在稳态下免疫系统和微生物组的变异情况,研究团队在旧金山湾区招募了110名健康参与者,建立了免疫微生物组队列(图1A)。参与者主要为非西班牙裔白人和亚裔成年人(图1B)。他们收集了血液和粪便样本,并进行了与一般健康状况、饮食和既往病史相关的调查。他们对外周血单核细胞(PBMC)进行了免疫谱分析,定量了血浆中的循环因子,生成了从粪便中提取的DNA的微生物组谱数据,并进行了粪便代谢组分析(图1A)。他们对每个数据集进行了预处理,以提取分子和细胞特征,并评估了这些特征在整个队列中的分布情况。
他们利用单细胞质谱流式细胞术(CyTOF)评估了免疫细胞群体的频率及其在队列中的变异。基于关键标记物的表达对细胞进行无监督聚类,并在力导向图上可视化了其相对丰度的变化(图1C)。虽然大多数细胞聚类在不同个体间存在差异(图1C),但根据变异系数对细胞聚类进行排序后发现,某些NKT、NK和T细胞聚类的变异程度最高(图1D)。
他们利用细胞转录组和表位测序索引(CITE-seq)技术评估了队列中免疫细胞亚群基因和蛋白表达的变异情况。基于细胞外蛋白表达和基因表达,对单个细胞进行聚类和注释,从而识别出包含已确定的免疫细胞类型的细胞聚类(图1E)。对每种细胞类型的基因表达矩阵进行了独立的降维处理,以评估队列中主要的转录变异轴。例如,在单核细胞中,可视化了队列中前10个维度上的基因表达变异情况(图1F)。维度1与参与干扰素(IFN)反应的基因呈正相关,但与参与炎症反应的基因呈负相关(图1G)。在其他细胞类型中也发现了类似规模的基因表达变异。
同时,他们利用宏基因组测序技术研究了微生物组组成的变化,鉴定了不同分类水平的微生物分支。正如预期,大多数鉴定出的物种属于4个主要门,并且在不同人群中表现出多样性差异。虽然丰度最高的5个科覆盖了大多数研究参与者已知分类单元的50%以上,但每个科的相对丰度在不同个体间差异很大(图1 H)。这种组成上的差异在物种水平上也可见。这些观察结果与人类微生物组计划中发现的主要分类单元和丰度变化基本一致,该计划对更大规模的健康人群进行了采样。
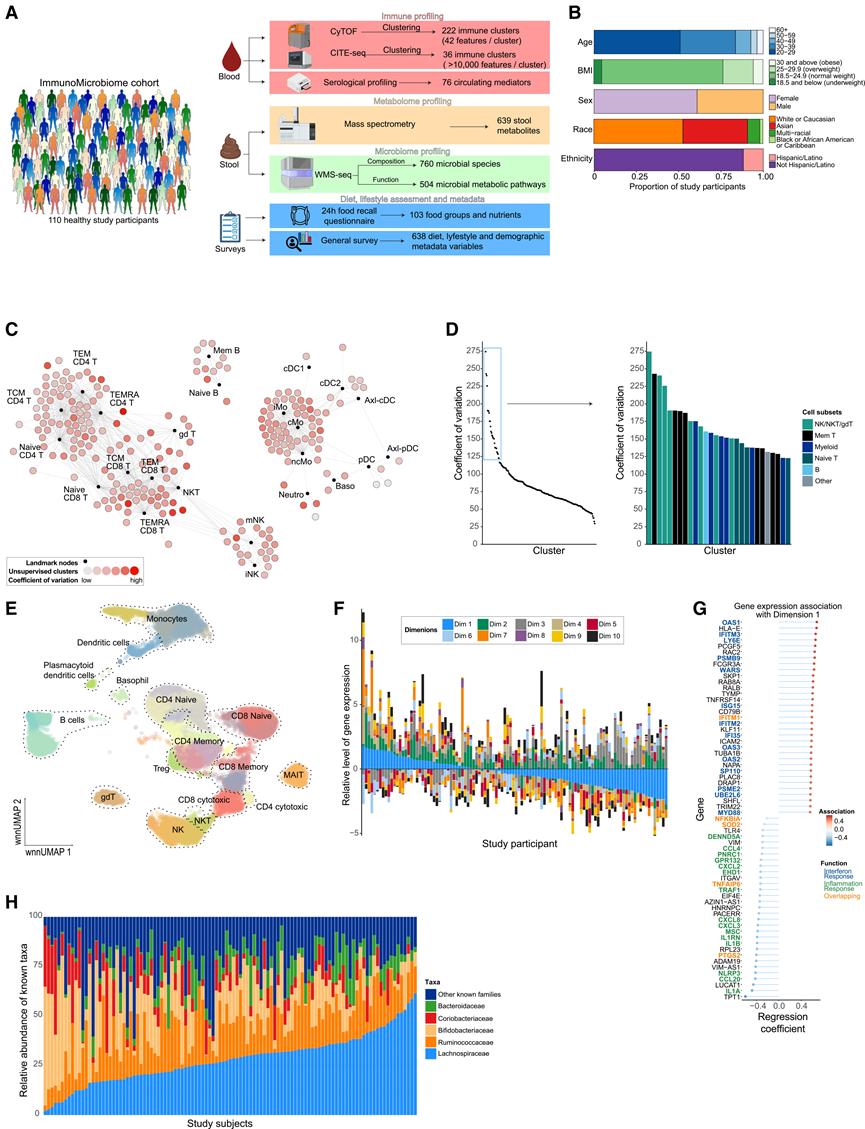
图1. 健康人群队列中免疫和微生物组的变异情况。
(A) 免疫微生物组队列概述和数据生成示意图。(B) 队列人口统计细分。(C) CyTOF 数据的支架图。(D) 左图:根据队列中变异系数从高到低对CyTOF聚类进行排序。右图:以柱状图形式展示了变异性最高的聚类。(E) CITE-seq 数据的 UMAP 可视化。(F) 队列中单核细胞的转录变异。(G) 单核细胞中与维度 1 显著相关的基因。(H) 研究参与者中最具代表性的 5 个微生物组家族的相对丰度。
02
整合多组学分析揭示了微生物组相关的IFN和炎症反应免疫变异
为了识别免疫系统和微生物组系统之间同时存在的生物学特征差异,他们采用多组学方法整合了不同模态的数据。他们应用多组学因子分析(MOFA)生成因子,这些因子由跨模态的协调特征组成,能够捕捉队列中不同的生物学变异。使用线性模型鉴定了与每个因子显著相关的特征,并评估了每个模态中与每个因子相关的特征的数量和比例(图2 A)。虽然因子(Factor)1捕捉到了免疫细胞基因表达的显著变异,但它与粪便微生物组或代谢组特征没有显著相关性(图2 A-B)。相比之下,因子3捕捉到了免疫系统、微生物组物种组成、微生物组通路丰度和代谢物的显著变异。在所有主要因素中,因子3在CyTOF和CITE-seq数据中均能最大程度地解释免疫细胞亚群相对丰度的变异。对物种水平微生物组组成数据进行主成分分析(PCA)发现,因子3与PC2之间存在显著相关性,CITE-seq基因表达数据的PCA也得出相同结论(图2C)。相比之下,因子1与CITE-seq基因表达主成分相关,但与微生物组组成主成分无关(图2D)。因此,他们将因子1称为独立免疫变异(IIV),将因子3称为免疫和微生物组伴随变异(IMCV)。
为了解IMCV所捕获的免疫变异,他们定量了不同细胞亚群转录数据中的主要特征。干扰素α (IFN-α) 反应、干扰素γ (IFN-γ) 反应和炎症反应的特征评分最高(图2E)。随后,确定了每个因素的主要标志性特征,并定量了富集这些特征的细胞亚群数量。在大多数免疫细胞群体中,IFN-α、IFN-γ 和炎症反应特征在IMCV中均显著富集(图2F)。有趣的是,这些特征也与IIV高度相关,IIV还捕获了其他特征(图2F)。这进一步表明,IFN和炎症反应是该队列中转录变异的主要来源。
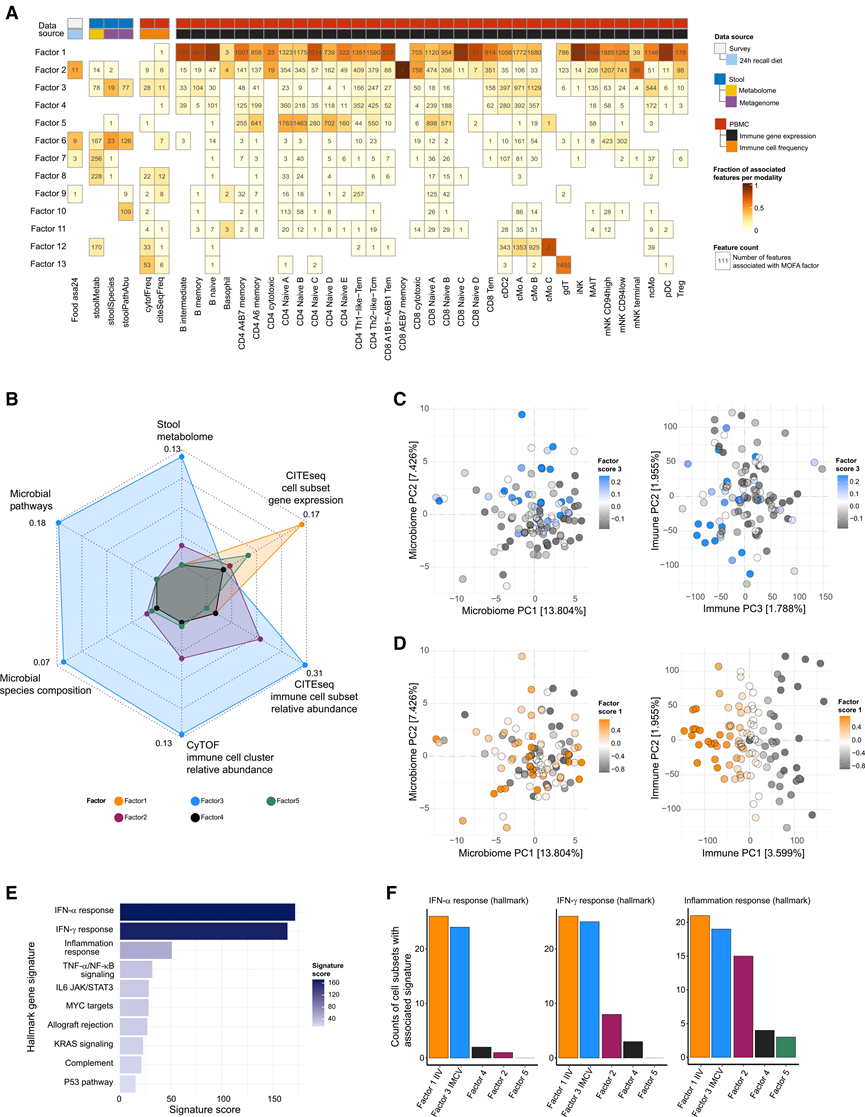
图2. 整合多组学分析揭示了微生物组相关的 IFN 和炎症反应的免疫变异。
(A) MOFA因子捕获的多组学特征。(B) MOFA因子按模态捕获的变异。(C-D) 流行微生物组物种组成数据(左)或 CITE-seq 基因表达数据(右)的PCA,按参与者的因子 3或因子 1 得分着色。(E) CITE-seq 数据中 IMCV 的转录特征富集在细胞亚群中的表现。(F)具有显著富集的标志性转录特征(IFN-α 反应、IFN-γ 反应和炎症反应)的细胞亚群数量。
03
IMCV 可以捕捉到队列中不同个体间 IFN 反应的差异
虽然IMCV 和 IIV 均广泛捕捉到了 IFN 相关特征(图 2F),但推测它们可能捕捉到了 IFN 反应的不同方面。使用与炎症相关特征不重叠的 IFN 基因集,他们观察到大多数细胞亚群在两种因子下均表现出 IFN 特征的正向富集,但大多数亚群与 IMCV 的关联更强(图 3A)。通过评估每个因子得分最高和最低(20%)的个体亚组之间细胞亚群的差异表达基因 (DEG),来研究驱动这些特征的具体基因。在高 IIV 亚组中,大多数上调基因在淋巴细胞亚群中表达(图3B-C),而在高 IMCV 亚组中,上调最显著的基因在髓系细胞亚群中富集(图3B-D)。大多数 DEG 仅存在于其中一个因子中(图 3E),IFN-α 反应特征也得到了类似的结果。
IFN信号通路会根据刺激的持续时间和强度诱导不同的转录反应。由于IMCV和IIV的IFN反应特征存在差异,他们研究了它们与更细致、更特异的IFN反应基因特征的重叠情况。与IMCV呈强正相关的基因富集于一种“持续性IFN反应”特征,该特征源自一项体内研究,该研究鉴定了对稳态组成型表达的IFN有反应的基因(图3F)。这些基因也富集于一种相关的“持续性IFN反应”特征,该特征由在体外人细胞中对持续低剂量IFN有反应而表达的ISG组成,这表明该队列中IFN暴露可能存在差异。相比之下,IIV与这些特征之间的关联要弱得多(图3F)。
因此,他们假设高IMCV亚组研究参与者的循环IFN水平可能更高。他们检测了血浆样本中的细胞因子水平,发现高IMCV研究参与者的血浆中IFN-γ水平以及由ISG编码的趋化因子CXCL10和CXCL11水平更高(图3G-H)。这些分子的血浆水平也与不同细胞亚群的强直性IFN反应基因特征相关。
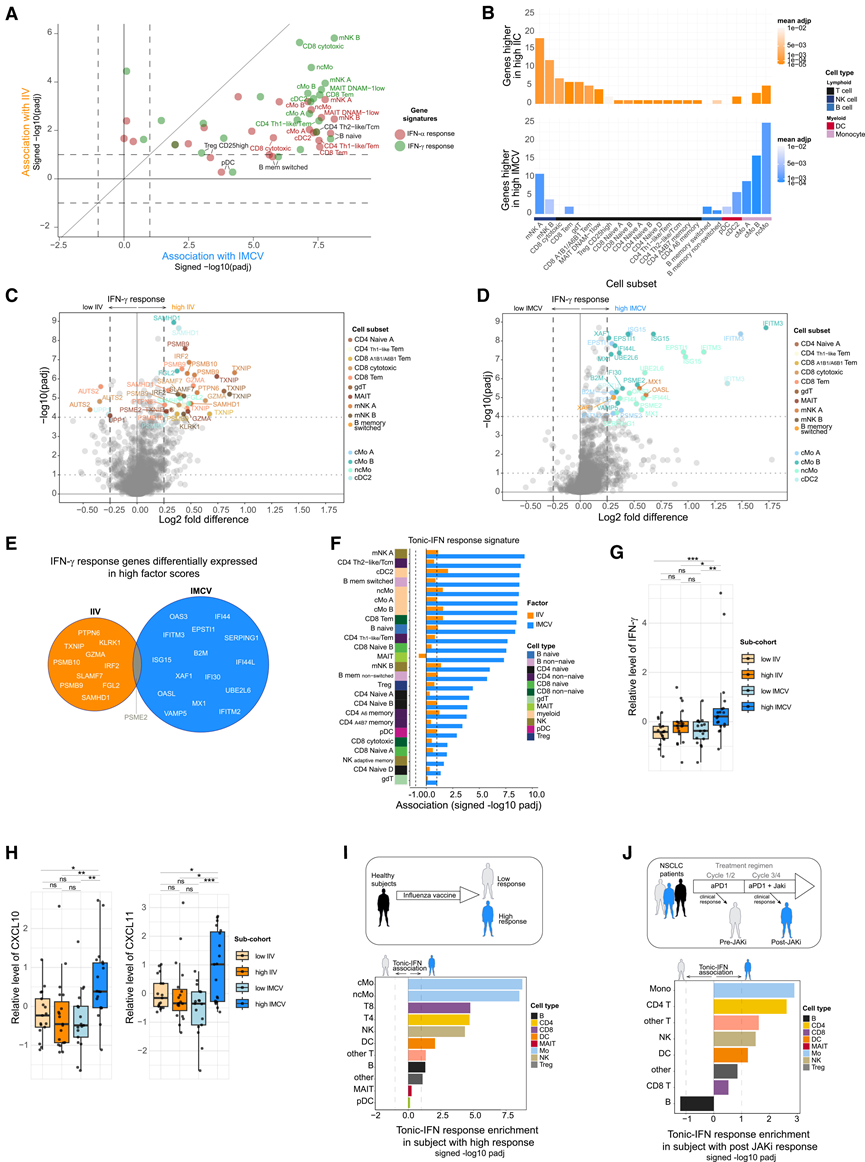
图3. IMCV 反映了队列中 IFN 反应的差异。
(A) 使用 mHG 测试分析 IIV 和 IMCV 与细胞亚群中 IFN 反应特征的关联。(B-E) 研究参与者中IFN-γ反应基因的数量和差异表达情况。(F) 使用 mHG 测试分析 IIV 和 IMCV 与细胞亚群中强直性 IFN 反应转录特征的关联。(G-H) 高 IIV 和 IMCV 因子评分与低 IIV 和 IMCV 因子评分的参与者血浆中 IFN-γ或 CXCL10 和 CXCL11的相对水平。(I) 在对季节性流感疫苗产生高反应或低反应的个体中,免疫细胞亚群中持续性干扰素基因特征的富集情况。(J) 在仅对PD-1单药治疗有反应或仅在与JAK抑制剂联合治疗后有反应的非小细胞肺癌患者的免疫细胞亚群中,tonic-IFN基因特征富集。
04
IMCV 和 IIV 之间的炎症和 TGF-β 转录程序受到不同的调控
之前的分析表明,炎症相关基因特征在整个队列中变异性也最高(图2E),并且与IMCV和IIV相关(图2F)。事实上,IIV和IMCV因子评分与IFN特征呈正相关,与炎症相关特征呈负相关(图4A)。在CITE-seq鉴定的3个主要单核细胞聚类中,IMCV和IIV均与炎症反应和肿瘤坏死因子α (TNF-α)/核因子κB (NF-κB)信号通路特征呈负相关(图4B)。然而,这些负相关性是由每个因子不同的差异表达基因(DEG)集驱动的(图4C-D)。IL-1通路基因,包括IL1A和IL1B,在IMCV评分高的个体中表达水平较低(图4C)。相比之下,高 IIV 组的 IL-6 调节因子表达较低,包括抑癌素 M (OSM)和TIMP1。TNF -α/NF-κB 信号通路特征也观察到类似结果(图 4D),其中高 IMCV 组中TLR/IFN 诱导的转录因子ATF3水平较高,而 ATF3 可抑制 NF-κB 信号通路。脂多糖 (LPS) 诱导的 TNF-α 因子 ( LITAF ) 可诱导 TNF-α 表达,其在高 IMCV 组中表达水平较高,但在高 IIV 组中表达水平较低(图 4D)。这些结果表明,IMCV和IIV均与单核细胞中的炎症和 TNF 通路呈负相关,但通过不同的基因集发挥作用。
在许多其他细胞亚群中,只有 IIV 与炎症相关特征呈负相关(图 4B)。MAIT 细胞是一类能够识别维生素代谢物(包括肠道微生物群来源的维生素代谢物)的不变T细胞亚群,是唯一 TNF-α/NF-κB 特征与 IMCV 呈正相关而与 IIV 呈负相关的亚群(图 4B)。驱动与 IIV 负相关的关键基因包括TNF、编码 NF-κB 亚基的基因及其调节因子(图 4E)。相反,与 IMCV 的正相关是由 TNF-α 下游炎症相关因子的高表达驱动的。转录因子JUNB(JUN 的负调控因子)在高 IIV 亚组中表达水平更高,提示 MAIT 细胞中 AP-1 通路的调控存在差异。虽然在其他淋巴细胞亚群中 TNF-α/NF-κB 通路与 IMCV 之间没有关联,但驱动 IIV 负相关的基因与 MAIT 细胞中的基因在很大程度上重叠(图 4 F)。
与这些结果一致,高IMCV和高IIV个体亚组的MAIT和成熟(m)NK-A淋巴细胞亚群的炎症基因特征均存在差异。TNF-α/NF-κB和炎症基因特征的综合评分也区分了高IMCV和高IIV亚组。由于微生物信号可以激活这些淋巴细胞亚群,他们还检测了激活标志物CD69的表面蛋白和mRNA表达。结果显示,高IMCV组的MAIT和mNK-A细胞中CD69的表达水平更高,导致CD69high细胞的相对丰度更高。
他们还研究了这些因素与TGFβ通路之间的关联,发现TGFβ通路与IIV呈独特的负相关(图4B)。高IIV组个体在多个细胞亚群中均表现出较低的TGFB1表达水平和较高的TGF-β受体抑制剂PPP1CA水平(图4G)。与IIV相反,cMo-A中的TGF-β特征与IMCV呈正相关(图4B),这主要是由于TGFB1、IFNGR2、JUNB、转录因子ID1和ID2的表达略有升高,以及参与TGFBR1抑制的PPP1R15A的表达降低所致(图4H)。
他们进一步评估了与这些通路相关的循环因子。单核细胞中的炎症和TNF-α/NF-κB信号通路特征与循环CD40水平呈正相关,CD40通过NF-κB通路发出信号(图4 I)。cMo-A中的TGF-β特征与血浆中潜伏期相关肽(LAP)-TGF-β1复合物水平以及其他与IMCV相关的因子(包括CXCL10和CXCL11)相关(图4 I)。与转录组数据一致,可溶性LAP-TGF-β1水平与IIV呈负相关(图4 J)。综上所述,这些数据表明,高IMCV和高IIV亚组在炎症和TGF-β信号通路方面存在关键差异。
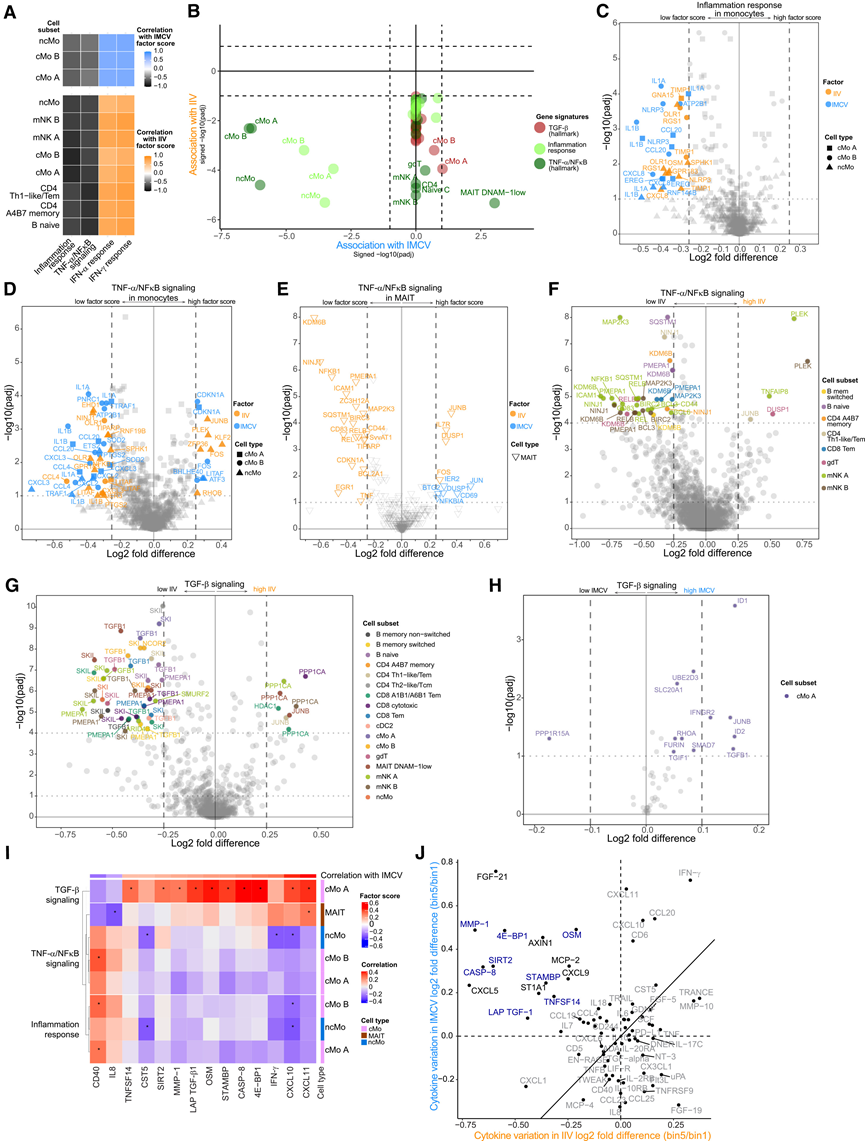
图4. 炎症和 TGF-β 转录程序在 IMCV 和 IIV 之间存在差异性调控。
(A) IIV 和 IMCV 因子评分与 IFN 和炎症相关特征(核心基因加权表达的总和)的相关性。(B) IIV 和 IMCV 与细胞亚群中炎症相关特征的关联。(C-E) 炎症反应基因或TNF-α/NF-κB信号通路基因在单核细胞亚群或MAIT细胞中,在因子评分高低不同的参与者之间差异表达。(F) TNF-α/NF-κB信号通路基因在IIV评分高低不同的参与者之间存在差异表达。(G-H) 与 F 相同,用于高 IIV 评分和低 IMCV 评分中的 TGF-β 信号特征。(I) 血浆细胞因子与IMCV及炎症相关特征在不同细胞亚群中的关联。(J) IIV 和 IMCV 中高因子评分和低因子评分之间的血浆细胞因子水平。
05
IMCV 能够捕捉到队列中 SIGLEC-1高表达单核细胞和 PD1高表达ICOS高表达记忆性 T 细胞的差异
IMCV 与 IIV 的区别还在于其与免疫细胞群丰度的关联(图 2A)。具有独特 IFN 和炎症转录特征的单核细胞亚群的相对丰度与 IMCV 呈正相关。CyTOF 数据由于其更高的通量,可以进行更深层次的采样,证实了几个单核细胞聚类的相对丰度与 IMCV 呈正相关(图 5A)。因此,他们将这些细胞聚类的蛋白表达与不相关的单核细胞聚类进行比较,以了解它们之间的差异(图 5B)。与 IMCV 相关的单核细胞聚类中,主要组织相容性复合体 (MHC) II 类蛋白 HLA-DR 和 Fcγ受体 CD64 的蛋白表达水平更高,而这两种蛋白均受 IFN 调控(图5B-C)。此外,这些细胞表达更高水平的髓系活化标志物(CCR7和CD14)以及与抗炎表型相关的清道夫受体 CD163(图5B-C)。CITE-seq 经典单核细胞聚类 B (cMo-B) 的丰度与 IMCV 相关,与其他 cMo 亚群相比,cMo-B 也表现出更高的 MHC II 类通路基因(HLA-DP/DQ/DR 和 CD74)表达。它们还表现出更高的免疫蛋白酶体基因PSME2和钙网蛋白 (CALR) 的表达,CALR 是一种对抗原加工和呈递至关重要的分子伴侣,以及 ISG(图 5D)。相反,cMo-B 中促炎基因的表达较低,包括 AP-1 亚基(JUN和FOS)以及内源性 TLR4 配体钙卫蛋白(S100A8 / S100A9)(图 5D)。为了进一步研究 IMCV 所反映的髓系细胞表型差异,他们比较了 IMCV高和低评分个体中 CITE-seq 蛋白的表达水平。SIGLEC-1 (CD169) 此前已证实可识别具有增强抗原呈递能力的 IFN 诱导的单核细胞,并可保护小鼠免受脓毒症或人类免受重症COVID-19的侵害,在 IMCV 高评分个体的单核细胞中表达更高(图5E)。虽然cMo-B细胞中SIGLEC-1的表达水平最高,但其他髓系细胞亚群在IMCV高表达组研究参与者中也表现出类似的差异表达,提示SIGLEC-1表达升高可能是这些个体髓系细胞状态更广泛特征的表现。与IIV高表达组相比,IMCV高表达组参与者的单核细胞中SIGLEC-1的表达水平也更高,导致SIGLEC-1高表达单核细胞的相对丰度更高。
多个记忆性T细胞聚类与IMCV呈正相关(图5A)。与未与IMCV相关的记忆性CD4或CD8 T细胞聚类相比,记忆性CD4 T细胞聚类115(C115)和CD8 T细胞聚类166(C166)表现出更高水平的共刺激和共抑制检查点分子组合表达(图5F)。这些细胞聚类还表达更高水平的激活标志物HLA-DR和转录因子Foxp3,以及更低水平的细胞因子受体CD127和CD25。另外两个与IMCV呈正相关的细胞聚类表现出类似的表型,只是Foxp3的表达有所不同(图5G)。与 Treg 细胞以及与 IMCV 无关的记忆 T 细胞相比,这四个细胞聚类的细胞表达 Foxp3 和大多数免疫检查点分子,包括抑制性免疫检查点配体 PD-L1,均较高。与 IMCV 呈负相关的 CD4 和 CD8 记忆 T 细胞聚类(图 5 A)通常呈现相反的表达谱。他们进一步分析了这些 CyTOF 细胞聚类的丰度与 CD8 效应记忆 T 细胞 CITE-seq 数据中的基因表达之间的相关性。大多数正相关基因与 T 细胞活化相关,包括许多抗原加工和呈递分子(图 5 H),这与 CyTOF 的结果一致。综上所述,这些结果表明 IMCV 与具有活化表型和免疫检查点分子高表达的记忆 T 细胞相关。
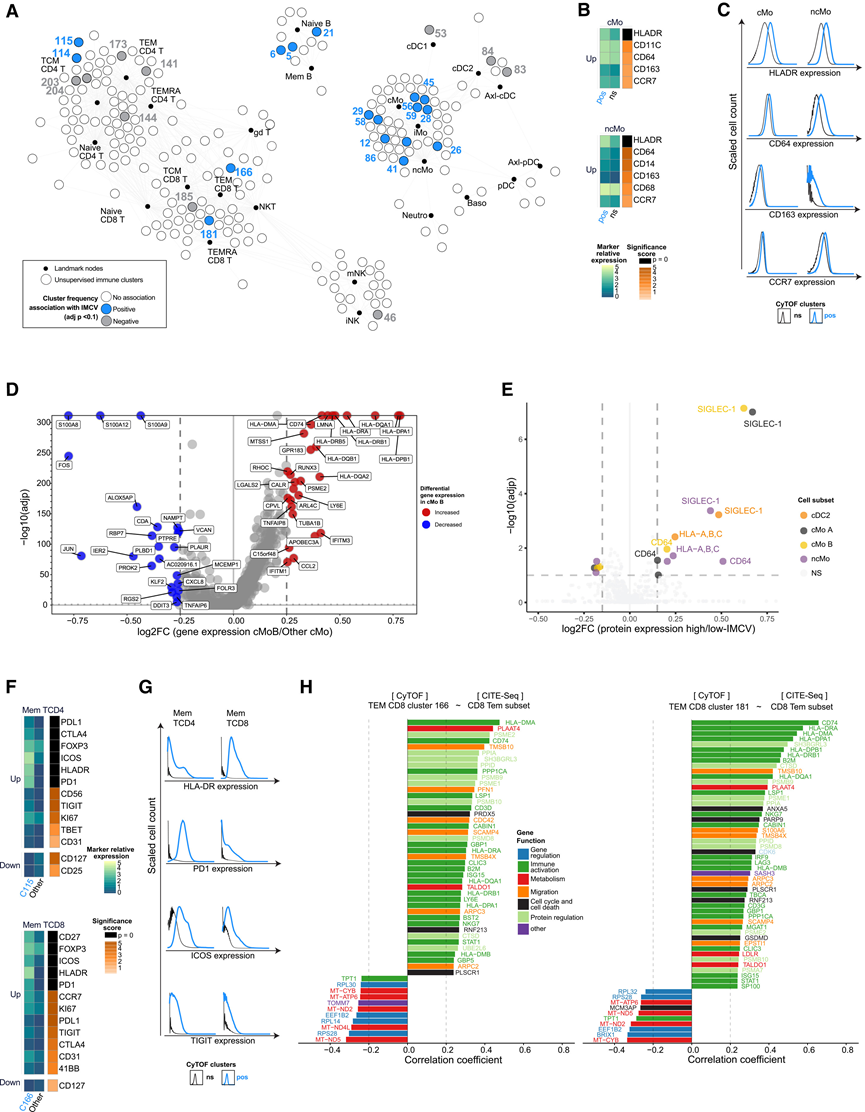
图5. 队列中 SIGLEC-1high单核细胞和 PD1highICOShigh记忆性 T 细胞的差异。
(A) CyTOF 聚类频率与 IMCV 关联的 SCAFoLD 图。(B) 上图:比较与IMCV显著正相关的经典单核细胞聚类(pos)和与IMCV无显著相关性的经典单核细胞聚类(ns)中CyTOF蛋白的表达情况。左图:蛋白表达值的中位数;右图:组间差异显著性;下图:非经典单核细胞聚类的相同结果。(C) 选定表面蛋白的表达。(D) 使用 Wilcoxon 秩和检验分析 cMo-B 与其他经典单核细胞亚群之间的差异性 CITE-seq 基因表达。(E)比较低 IMCV 因子评分和高 IMCV 因子评分的研究参与者之间的蛋白表达差异。(F) 与 B 相同,将记忆 CD4 T 细胞聚类 115 与所有与 IMCV 无关的记忆 CD4 T 细胞聚类进行比较,并将 CD8 T 细胞聚类 166 与所有与 IMCV 无关的记忆 CD8 T 细胞聚类进行比较。(G) 与 C 相同,但合并记忆 CD4 T 细胞聚类 114/115 和 CD8 T 细胞聚类 166/181。(H) CD8效应记忆T细胞中CITE-seq基因表达与CD8效应记忆T细胞聚类C166和C181的CyTOF频率之间的相关性。
06
已建立的免疫调节肠道微生物组通路和分子与 IMCV 相关
IMCV最独特的特征在于其免疫组和微生物组数据集之间的协调性(图2A)。结合MOFA的基于参考的分析方法鉴定出77条与IMCV相关的微生物通路,其中包括几条与已知免疫调节分子产生相关的通路:短链脂肪酸(SCFA)的生物合成、多胺的代谢以及细菌细胞壁成分脂多糖(LPS)和肽聚糖的产生或修饰(图6A)。此外,还有78种粪便代谢物与IMCV相关,其中包括这些通路产生的许多免疫调节分子(图6B)。SCFA乙酸、丁酸和丙酸以及SCFA相关化合物均与IMCV呈正相关。肌醇是丙酸的前体,与IMCV呈正相关,而其磷酸化前体肌醇-P则呈负相关。在检测到的8种多胺中,有6种与IMCV呈正相关,其中包括鸟氨酸,其前体与IMCV呈负相关。初级胆汁酸鹅脱氧胆酸和胆酸也因其免疫调节作用而受到研究,它们也与IMCV相关。综上所述,这些发现表明,IMCV所反映的免疫状态与肠道菌群及其代谢产物中的免疫调节通路相一致。
接下来,他们研究了编码这些代谢途径的细菌种类,通过统计它们在整个队列中携带的相关基因的丰度和平均拷贝数来评估它们对代谢途径丰度的贡献。长双歧杆菌(Bifidobacterium longum)、青春双歧杆菌(Bifidobacterium adolescentis)、假链状双歧杆菌(Bifidobacterium pseudocatenulatum)和Collinsella aerofaciens是乙酸生物合成途径的主要贡献菌种(图6C),它们的相对丰度也与乙酸生物合成途径的丰度相关。长双歧杆菌的丰度与IMCV直接相关。一些相同的细菌种类也是一系列代谢反应的主要贡献菌种,这些反应将乙酸转化为丁酸,将肌醇转化为丙酸(图6D)。其中,Blautia obeum和R. torques还编码了参与催化肌醇磷酸脱磷酸化的碱性磷酸酶基因家族,这表明这些细菌在丙酸生物合成中可能发挥着作用。
一组部分重叠的物种参与了与多胺代谢相关的通路(图 6E)。其中一些物种含有编码精氨酸转化为腐胺的基因,且这些基因的丰度较高。这些物种也是后续亚精胺和精胺生成的主要贡献者。B. adolescentis在精胺/亚精胺乙酰化相关基因家族的丰度中贡献最大,R. torques和B. obeum则是赖氨酸合成尸胺相关基因家族的重要贡献者。总体而言,参与多胺代谢的物种的互补性提示了群落代谢的存在。
通过对参与LPS生物合成的菌种(由先天免疫受体TLR4检测)的分析,揭示了更多重叠之处。双歧杆菌属和产气梭菌也是LPS前体N-乙酰氨基葡萄糖生物合成的重要贡献者(图6F)。它们的丰度与LPS生物合成途径的丰度进一步相关。N-乙酰氨基葡萄糖随后转化为LPS内毒素KDo2-脂质A的过程主要由特定菌种主导,包括大肠杆菌和副流感嗜血杆菌。这些途径中的几个主要贡献菌与IMCV呈正相关,其中一些与SCFA和多胺代谢途径的贡献菌重叠。这些菌种中的一些也是胆盐水解酶基因家族丰度的主要贡献者,该基因家族能够产生与IMCV相关的非结合型胆汁酸(图6B)。
携带参与 IMCV 相关分子(短链脂肪酸、多胺、脂多糖、初级胆汁酸)合成基因的物种具有系统发育多样性,涵盖超过 14 个科。一个核心物种群落编码参与所有 IMCV 相关关键过程的基因,涵盖 5 个微生物科。值得注意的是,这些微生物编码的代谢途径和代谢物水平与 IMCV 的相关性强于细菌物种的丰度。关键微生物组途径丰度和粪便代谢物的 IMCV 微生物组综合评分与免疫细胞亚群的强直性 IFN 反应特征显著相关(图 6 G)。这些结果凸显了 IMCV 捕获的免疫调节分子合成途径所涉及的复杂系统发育多样性微生物群落。
与基于参考基因组的宏基因组数据分析方法相比,基因组解析分析能够更精确地识别具有不同功能的菌株。他们对去除人类基因组的宏基因组进行了从头组装,以识别宏基因组组装基因组(MAG),并由此构建了亚种基因组库(SGB)。然后,将每个宏基因组映射到SGB库,构建基因组解析的群落计数矩阵,并使用基因组分类数据库(GTDB)对每个SGB进行分类注释,以确定其最接近的物种。根据Spearman相关性距离对SGBs进行分组,最终得到60个共丰度SGB模块。在这些共丰度模块中,有7个与IMCV显著相关(图6H)。在与IMCV呈正相关的3个模块中,模块C捕获了31个SGB,反映了Collinsella属内遗传上不同的菌株,其中7个与多个C. aerofaciens参考菌株特异性匹配(图6I)。模块F包含4个SGB,与多个双歧杆菌参考菌株(长双歧杆菌、链状双歧杆菌、假链状双歧杆菌和青春双歧杆菌)以及多个与Phocaeicola plebeius匹配的SGB相匹配(图6I)。这些发现通过识别与IMCV丰度直接相关的SGB,强化了基于参考菌株的通路贡献分析方法所鉴定的细菌群落。基因组解析分析还发现了一个与 IMCV 评分显著正相关的附加模块,即模块 K,其中包括与Turicibacter bilis、Clostridium saudiense、Romboutsia timonensis和Intestinibacter bartlettii相关的 SGB (图6I)。
在与IMCV呈负相关的模块中,模块M主要由几种Alistipes SGB组成(图6J)。在之前的分析中,Alistipes putredinis已鉴定为几种IMCV相关通路的主要贡献者,在这些SBG中,它与IMCV的相关性最强(图6J)。通过基因组解析方法独特地鉴定出几个与IMCV呈负相关的模块,包括模块H,其中包含与Lachnospiraceae相关的Eubacterium_F sp003491505_3和Eubacterium_F sp003491505_1等;模块S,其中包含与Gallintestinimicrobium propionicum、Acetatifactor intestinalis、Acetatifactor acetigignens和Roseburia hominis相关的SGB;以及模块U,其中包含几种Hominicoprocola菌株(图6J)。总体而言,来自 IMCV 相关模块的各个 SGB 的相对丰度与持续性 IFN 反应特征以及关键 IMCV 通路和代谢物的相关性表现出一致的方向性(图6K)。
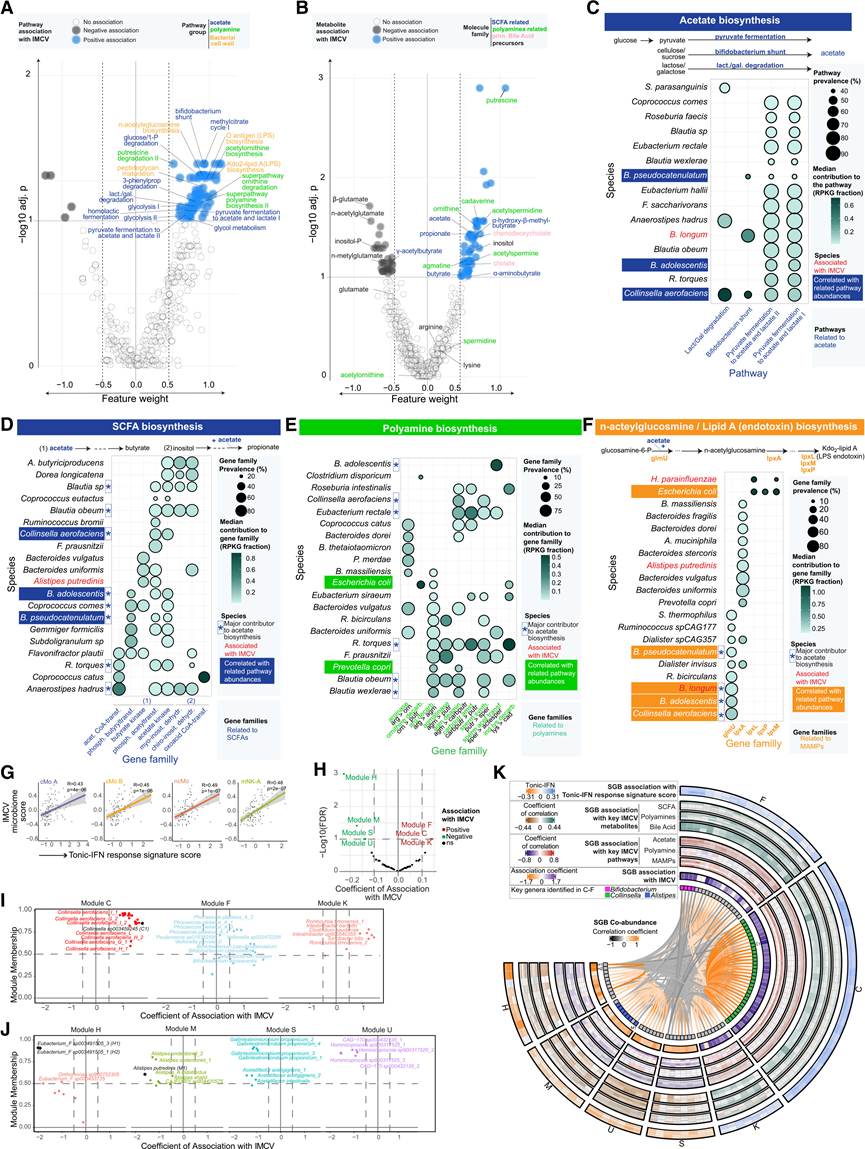
图6. 免疫调节肠道微生物组通路和分子与 IMCV 相关。
(A) 微生物组通路丰度与IMCV之间的关联。(B) 粪便代谢物与IMCV的关联。(C) 各物种对乙酸生物合成途径基因组内容的贡献。(D-F) 物种对短链脂肪酸(SCFA)生物合成、多胺代谢以及Kdo2-脂质A[LPS-内毒素,一种微生物相关分子模式(MAMP)]生物合成相关基因家族基因组含量的贡献。(G) 微生物组综合评分与强直性 IFN 反应基因特征之间的 Spearman 相关性和线性回归线。(H) IMCV 与 SGB 共丰模块频率之间关联的火山图。(I-J) 散点图显示了 SGB 模块成员得分和与 IMCV 相关的关联系数。(K) Circos 图可视化了与 IMCV 和感兴趣的代谢通路/代谢物相关的模块中 SGB 的共丰度。
07
IFN 反应特征和 IMCV 相关特征随时间推移保持稳定
他们假设IMCV的特征在个体内部可能随时间保持稳定,反映了免疫系统和微生物群的协调设定点。因此,他们在基线采样时间点后约20个月的远期随访时间点,从队列的一个子集中收集了生物样本,并生成了一个纵向多组学数据集,以匹配之前的分析。该随访队列涵盖了整个免疫微生物组队列中观察到的所有IMCV评分范围。
为了评估免疫表型随时间的稳定性,他们对纵向血液样本进行了CITE-seq、血浆细胞因子和CyTOF分析。CITE-seq数据用于量化所有主要细胞类型的基础性IFN反应特征以及IFNα和IFNγ反应特征(图7A-B)。事实上,在先前观察到具有强烈IFN相关基因表达程序的关键细胞群中,不同时间点之间存在显著相关性(图3A-F),表明这些IFN反应特征在个体参与者中随时间推移保持稳定。同样,血浆中IFNγ和ISG编码的趋化因子CXCL10和CXCL11的水平在两个时间点之间也与参与者显著相关(图7C-D),CCL20和CD40的水平也与IMCV显著相关,而CCL20和CD40的水平在原始分析中也与IMCV显著相关。 CyTOF 分析显示,先前鉴定的关键免疫细胞亚群的频率(图 5A)在两个时间点之间也与所有参与者高度相关(图7E-F)。总的来说,这些结果表明,在一年多的时间里,导致 IMCV 的关键免疫参数在研究参与者体内相当稳定。
同时,为了研究微生物组及其代谢产物随时间的稳定性,他们对这些受试者的纵向粪便样本进行了宏基因组学和代谢组学分析。与IMCV显著相关的多数关键通路(图6A)在每位受试者的两个时间点之间均表现出高度相关性(图7G-H)。与IMCV相关的关键细菌类群的相对丰度也呈现同样的趋势。此外,与IMCV相关的关键代谢产物(图6B)在每位受试者的两个时间点之间也高度相关(图7I-J)。综上所述,这些结果表明与IMCV相关的微生物代谢通路和代谢物水平具有高度稳定性。
最后,他们对每位研究参与者在每个时间点的这些特征进行了降维,并通过均匀流形逼近和投影(UMAP)进行可视化(图 7K)。事实上,任何给定个体的两个时间点的数据都非常接近,表明这些免疫和微生物组相关特征随时间推移具有稳定性。
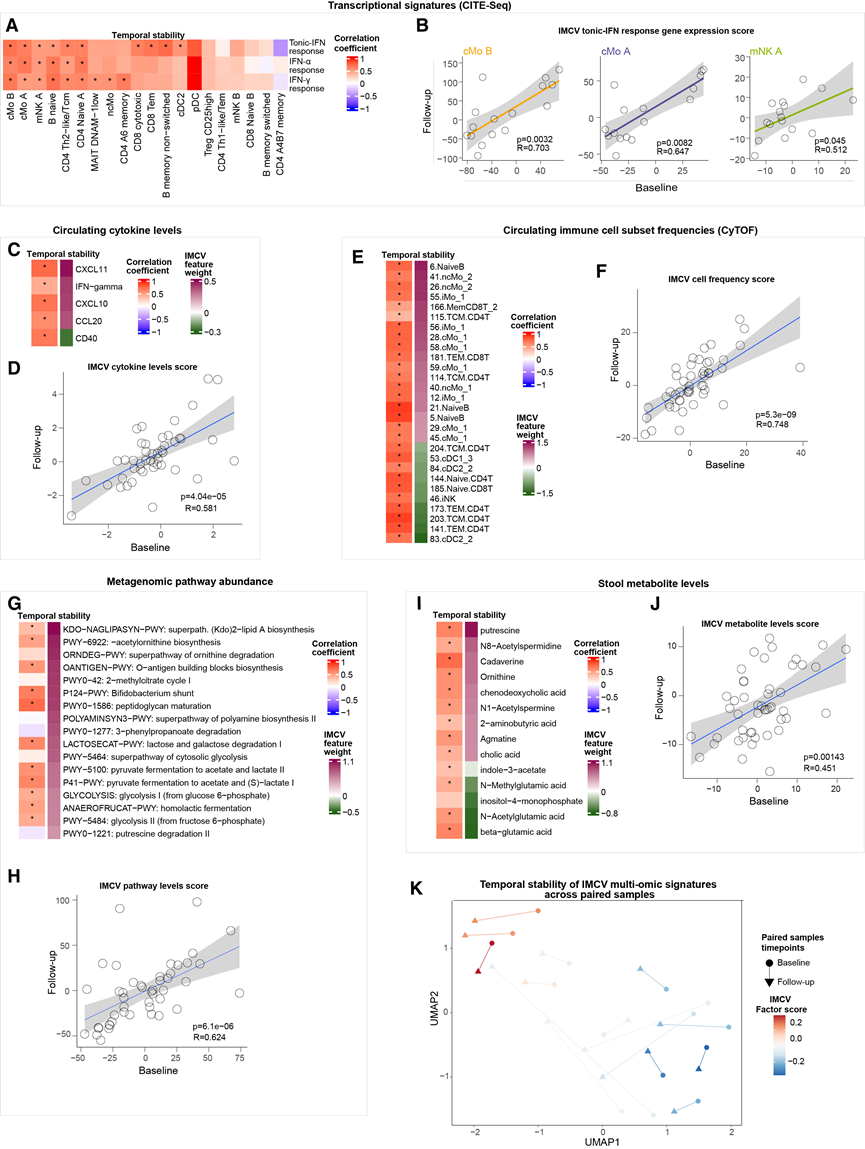
图7. IFN 反应特征和 IMCV 相关特征随时间推移保持稳定。
(A) 每位参与者 ( n = 16)在两个时间点之间,免疫细胞亚群中 IFN 相关基因特征的 Spearman 相关性。(B) 散点图可视化了关键细胞群在两个时间点的强直性干扰素反应基因表达评分。(C) 每个参与者在两个时间点之间与 IMCV 相关的循环可溶性因子的 Spearman 相关性。(D) 散点图可视化了两个时间点之间与IMCV相关的可溶性因子综合评分。(E) 采用CyTOF质谱法检测每位研究参与者在两个时间点与IMCV相关的细胞亚群相对丰度。(F) 散点图可视化了两个时间点之间与IMCV显著相关的细胞聚类的综合得分。(G) 每个研究参与者在两个时间点之间粪便宏基因组测序数据通路丰度的Spearman相关性。(H) 散点图可视化了两个时间点之间与IMCV显著相关的通路综合评分。(I) 每个研究参与者(n = 48)在两个时间点之间粪便代谢物丰度的 Spearman 相关性。(J) 散点图可视化了两个时间点之间与IMCV显著相关的代谢物综合评分。(K) 对上述所有关键特征在每个时间点进行UMAP降维。
+ + + + + + + + + + +
结 论
研究团队通过免疫微生物组研究对110名健康参与者进行了多组学分析。基于因子的整合方法识别出协同变异,揭示干扰素反应是健康参与者中最具变异性的免疫特征之一。微生物群组成、通路和粪便代谢物与干扰素反应通路同步变化。超过一年的纵向数据显示,这些参数在个体内部具有显著的稳定性。本研究提供了大量数据,可用于研究健康个体在稳态下的免疫状态与微生物群之间的关系,这为阐明与疾病易感性和治疗反应相关的个体差异奠定了基础。
+ + + + +



