

 English
English文献解读|Neuro Oncol(13.4):弥漫性中线胶质瘤的蛋白质组学图谱凸显了非组蛋白甲基转移酶的治疗潜力
✦ +
+
论文ID
原名:The proteomic landscape of diffuse midline glioma highlights the therapeutic potential of non-histone protein methyltransferases
译名:弥漫性中线胶质瘤的蛋白质组学图谱凸显了非组蛋白甲基转移酶的治疗潜力
期刊:Neuro-Oncology
影响因子:13.4
发表时间:2025.02.15
DOI号:10.1093/neuonc/noaf033.
背 景
过去十年的测序研究发现,组蛋白H3.3或H3.1中赖氨酸27由甲硫氨酸(K27M)取代的现象高度复发,主要局限于中线胶质瘤。这些肿瘤在儿童中尤为常见,最常发生于脑桥,传统上称为弥漫性内生性脑桥胶质瘤。随着H3K27M突变的发现,这类肿瘤在成人和其他中线区域(如丘脑或脊髓)的发生率也越来越高。鉴于此,世界卫生组织最新的中枢神经系统肿瘤分类已将这类肿瘤归类为弥漫性中线胶质瘤(DMG),H3K27突变型。DMG是一种无法治愈、侵袭性极强、弥漫浸润性脑肿瘤。目前临床上迫切需要有效的DMG治疗方案,现有疗法(如放疗和化疗)的局限性凸显了探索针对这种毁灭性疾病的新型治疗策略的必要性。虽然对DMG已进行了全面的基因谱分析,但对其蛋白质组和翻译后修饰(PTM)水平的研究仍然有限。
实验设计
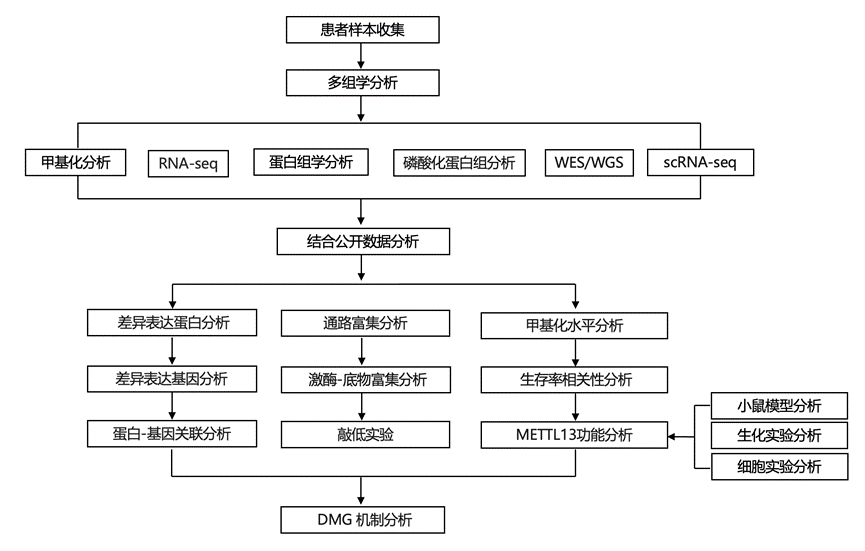
结 果
01

DMG的多组学分析
研究团队使用液相色谱-串联质谱(LC-MS)技术对 55 个患者样本(39 个 DMG 和 16 个正常脑组织)进行了深度总蛋白质组学分析,并进行了相应的转录组分析(RNA-seq)、全基因组测序 (WGS)、全外显子组测序 (WES)和甲基化分析(图 1a)。对 30 例患者样本进行了磷酸化蛋白质组和甲基化蛋白质组分析。本研究的队列包括 21 名女性和 18 名男性,诊断时的中位年龄为 6.8 岁,中位生存期为 0.8 年。
使用主成分分析,DMG 样本在转录组、蛋白质组和 PTM 水平上与正常脑组织明显不同。他们在 DMG 和正常脑组织之间鉴定出 3152 个差异表达蛋白(占已鉴定 9530 个总蛋白的 33%)和 7880 个差异表达 RNA(占已鉴定 35722 个 RNA 的 22%)(编码和非编码 RNA)。DMG 中差异表达蛋白的通路富集分析显示,细胞粘附、翻译、GTPase 活性和致癌 MAPK 信号通路富集,而膜运输、TCA 循环和神经系统发育则呈负富集(图1B)。
mRNA和蛋白质的相关性在理解基因表达调控及其对细胞功能的影响方面发挥着至关重要的作用。通常发现mRNA和蛋白质水平之间的相关性为中等。然而,差异表达的mRNA与蛋白质的相关性显著高于非差异表达的mRNA。与此相符的是,在3059个差异表达的DMG匹配的蛋白质:mRNA对中,蛋白质和mRNA水平之间存在中等至较高的相关性。然而,有 395 个显著上调的蛋白质在 mRNA 水平上没有差异表达,因此在转录组学研究中会遗漏。这些蛋白质具有主要功能,包括 RNA、蛋白质和维生素的代谢,以及表观遗传调控、DNA 修复和去泛素化。这种差异可能源于翻译后调控,包括PTM、蛋白质降解和翻译效率,这些因素都会独立于 mRNA 丰度影响蛋白质水平。因此,蛋白质组学提供了一种正交且更直接的细胞过程理解方式,使大众能够发现仅通过 mRNA 分析无法发现的重要调控机制。
与正常脑组织相比,DMG 中共有 218 种蛋白质(占 448 种已鉴定磷酸化蛋白质的 47%)的磷酸化水平存在差异,其中 102 种蛋白质的磷酸化水平升高,116 种蛋白质的磷酸化水平降低(图 1C)。DMG 中差异磷酸化蛋白的通路分析显示,mRNA 剪接、Rho GTPase 信号通路、MAPK 信号通路、细胞周期和翻译起始呈正富集(肿瘤中高磷酸化蛋白),而膜转运、信号转导和小分子运输呈负富集(图 1C)。肿瘤中磷酸化不足的蛋白,例如 MADD、DCTN1 和 SLC4A10,在未磷酸化时会激活通路或促进肿瘤发生。
在 218 个差异磷酸化蛋白中,有 80 个在 DMG 中没有差异表达,这表明这些蛋白只能通过观察 PTM 来检测,而不能通过观察表达来检测。参与蛋白质折叠、蛋白质代谢、糖基化和自噬途径的 PTM 富集蛋白质在其表达水平(蛋白质或 mRNA)上并未富集(图 1D)。
对DMG中磷酸化蛋白的定量分析显示,与正常脑组织相比,DMG中平均整体蛋白磷酸化水平显著降低(图1E)。这主要是由于丝氨酸和苏氨酸磷酸化水平降低所致,而酪氨酸磷酸化水平(包括致癌磷酸肽)则显著升高(图1F)。虽然既往研究报道癌症中整体酪氨酸磷酸化水平升高,但此前尚未报道癌症患者组织中整体丝氨酸/苏氨酸磷酸化水平降低。与正常脑组织相比,DMG中共有29种磷酸酶的蛋白表达水平显著升高。虽然肿瘤抑制性磷酸酶 PP2A、PPP2R5E 和 PPP2CB(其底物 VIM 在 DMG 中高度磷酸化)的亚基在 DMG 中下调,但调节细胞分裂和 DNA 修复的致癌性磷酸酶,如 PPP6R1 和 PPP4R1,在 DMG 中上调DEPOD。观察到的磷酸化水平降低主要归因于肿瘤发生,特别是参与膜转运和信号转导通路的蛋白质(图 1C)。例如,某些蛋白质(如参与溶酶体转运和自噬的 DCTN1)磷酸化水平降低,可激活其功能活性。类似地,MADD 蛋白磷酸化后与 TRAIL 相互作用诱导细胞凋亡;因此,MADD通过去磷酸化受到负调控,从而促进肿瘤发生。此外,去磷酸化有助于 SLC4 蛋白(特别是小分子转运蛋白)的稳定。值得注意的是,该蛋白家族成员 SLC4A10 在 DMG 组织中的磷酸化水平显著低于正常脑组织,提示其可能对小分子转运过程产生影响。
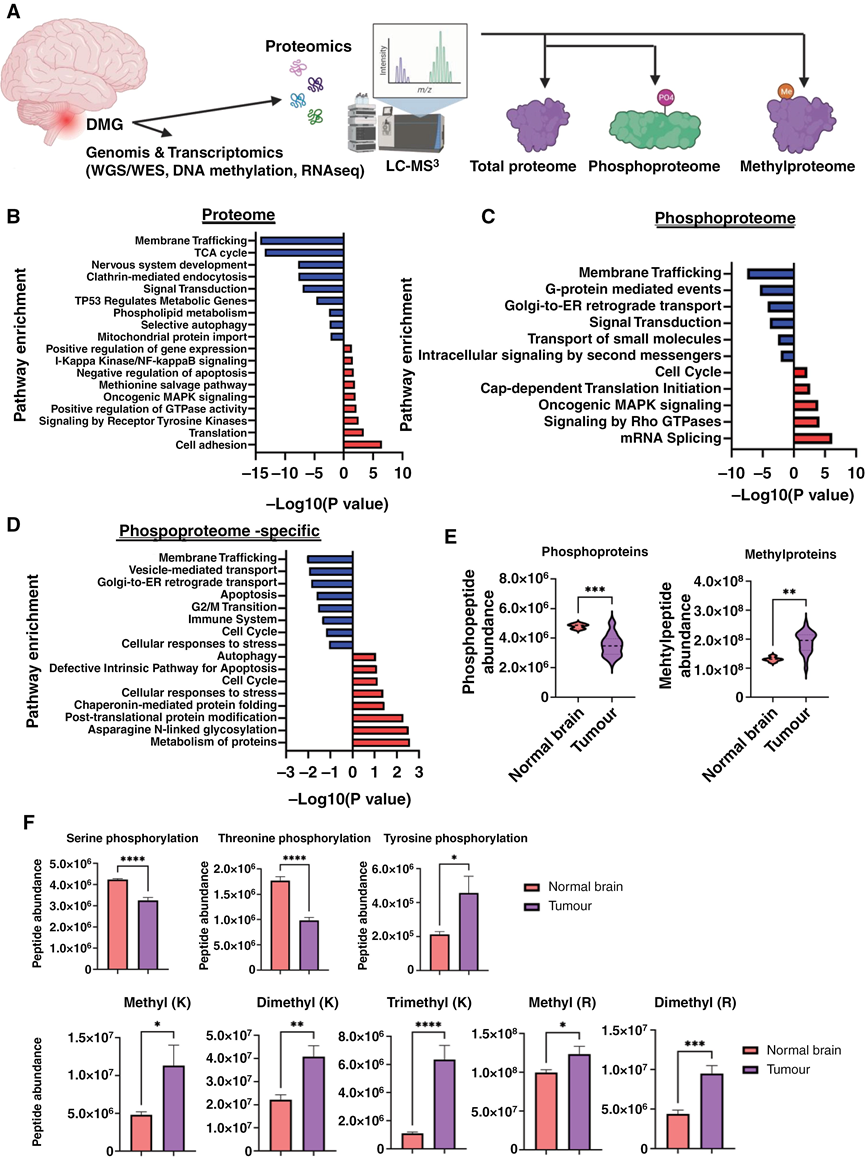
图1. DMG患者样本的多组学分析。
(A) DMG样本的多组学分析流程,包括全基因组测序/全外显子组测序(WGS/WES)、DNA甲基化、RNA-seq、总蛋白质组、磷酸化蛋白质组和甲基化蛋白质组。(B) DMG中差异表达蛋白的通路富集分析。(C) DMG中差异磷酸化蛋白的通路富集分析。(D) DMG中差异表达但磷酸化程度不同的蛋白的通路富集分析。(E) 正常脑组织和DMG组织中蛋白质的整体磷酸化和整体甲基化水平。(F) DMG中丝氨酸、苏氨酸和酪氨酸的整体磷酸化水平,以及DIPG中赖氨酸和精氨酸的整体甲基化水平及其各种修饰。
02
PRKACA、PAK2 和 AKT1 是 DMG 磷酸化蛋白质组的关键驱动因子
为了揭示DMG磷酸化蛋白质组的关键驱动因素,他们进行了激酶-底物富集分析(KSEA)。基于底物特定肽序列上差异磷酸化的S/T/Y氨基酸的差异倍数,计算了每种激酶的25个KSEA富集评分。KSEA鉴定出PRKACA、PAK2和AKT1是DMG中差异磷酸化蛋白的主要驱动激酶(图2)。为了进一步验证DMG磷酸化蛋白质组的关键驱动因素,他们对3种DMG细胞系(SU-DIPGXVII、SU-DIPG25和SU-DIPG50)进行了全面的磷酸化蛋白质组分析,鉴定出1323个独特的磷酸化肽,对应于600个独特的磷酸化蛋白。为了探索驱动 DMG 细胞中磷酸化的信号通路,他们使用 KEA3 进行了KSEA。该分析揭示了总共 51 个显著富集的激酶,包括关键调节因子,例如 CDK1、CDK2、PAK 家族、AKT1、PRKACA、mTOR 和 AURKA。细胞周期调控的磷酸化蛋白,例如 CDK1 和 CDK2,受到高度调控且去磷酸化迅速,因此可能在人类样本中遗漏;然而,在 DMG 肿瘤组织中高度磷酸化的 AKT1 和 PRKACA 的底物,在 DMG 细胞系中也表现出高磷酸化水平。
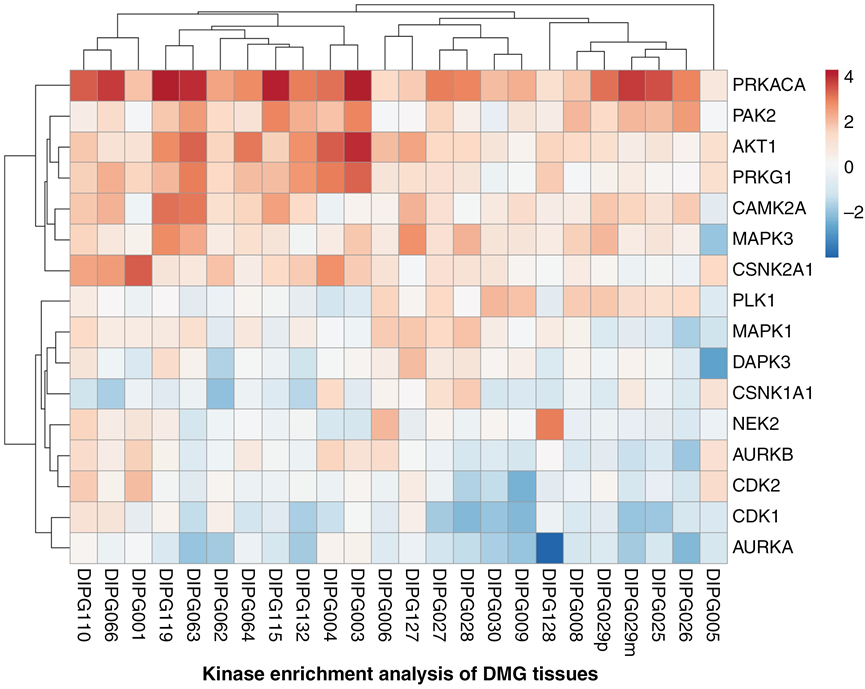
图2. DMG中的蛋白质磷酸化。基于DMG组织中底物磷酸化情况,通过激酶富集分析鉴定出的主要蛋白激酶。
接下来,他们希望研究这些激酶作为DMG治疗靶点的潜力。AKT1抑制此前已证明可以抑制DMG细胞生长;然而,靶向PRKACA和PAK2的潜力尚不清楚。他们使用两种独立的shRNA敲低了PRKACA和PAK2,并通过质谱和Western blot在蛋白水平上证实了敲低(图3A)。敲低 PRKACA 和 PAK2 蛋白显著降低了 SU-DIPGXVII 和 SU-DIPG36 细胞的生长(图 3B)。然后,他们使用质谱分析鉴定了 PRKACA 和 PAK2 敲低的 SU-DIPGXVII 细胞中差异表达的蛋白质,并将其与对照 scrambled-ShRNA 进行比较。通路富集分析突出显示,翻译和细胞周期是 PRKACA 和 PAK2 敲低细胞中受影响最大的通路(图 3C)。PRKACA 和 AKT1 主要在恶性细胞中表达,而 PAK2 也存在于免疫细胞和少突胶质细胞群中表明,靶向 PAK2 可能导致脱靶毒性,而 PRKACA 和 AKT1 可能是更具选择性的靶点。AKT1 是 PI3K/AKT/mTOR 通路的关键效应分子,该通路在弥漫性胶质瘤 (DMG) 中经常出现异常表达,并与细胞生长、存活和耐药性有关。
PRKACA(PKA,催化亚基)是cAMP依赖性信号通路的核心调节因子,控制着包括转录、代谢和细胞凋亡在内的多种生物学过程。PAK2是细胞骨架动力学、细胞存活和迁移的重要介质,已知其与RAS和MAPK通路相互作用。这些通路与H3K27M驱动的DMG密切相关,其中RAS/MAPK信号通路支持肿瘤生长和耐药机制。在DMG中,他们鉴定的PRKACA底物主要调控转录后调控蛋白,而AKT1和PAK2的底物则主要影响翻译起始复合物的组成成分,以及这些激酶磷酸化的其他经典蛋白。
目前尚无直接靶向 PRKACA 的药物;然而,可以通过靶向 PKA来间接抑制 PRKACA,PRKACA 是 PKA 全酶的催化亚基。为了探究这一点,他们检测了 H-89 和 A443654(分别抑制 PKA 和 AKT1 活性)在 DMG 细胞中的活性。两种药物均能显著抑制 DMG 细胞的生长,其IC50 值与其他有效肿瘤的 IC50 值一致(A443654 在 SU-DIPG36 和 SU-DIPGXVII 中的 IC50 值分别为 130 nM 和 180 nM;H-89 在 SU-DIPG36 和 SU-DIPGXVII 中的 IC50 值分别为 7 μM 和 3.1 μM)。随后,他们将两种药物以其各自的 IC50浓度联合使用,观察到与单独使用任一药物相比,对 DMG 细胞生长的抑制作用显著增强(图 3D)。这些结果与基于 shRNA 的基因敲低实验结果一致(图 3A-B),进一步证实了这些激酶在 DMG 中的关键作用,并表明它们是药物干预的可行靶点。
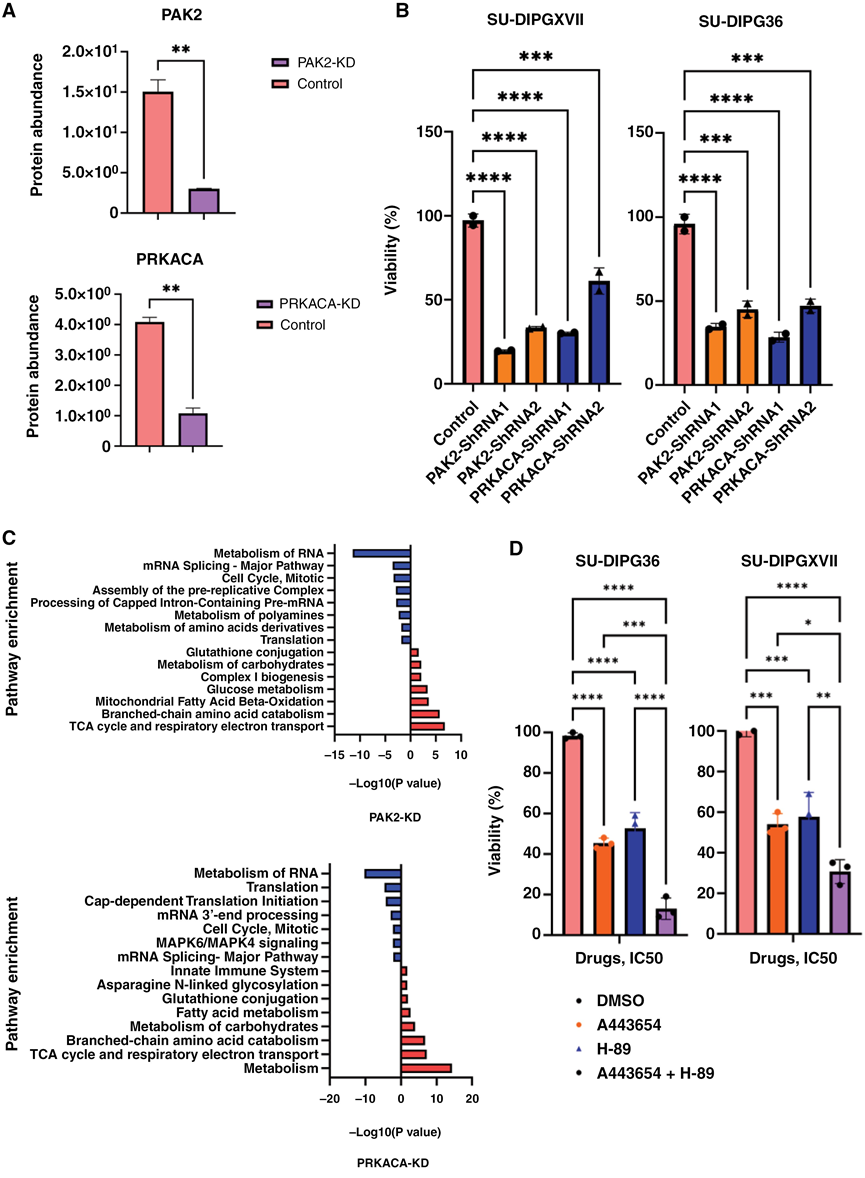
图3. 蛋白激酶候选物在DMG中的作用。
(A) 质谱分析证实了基于shRNA的DMG(SU-DIPGXVII)细胞中PAK2和PRKACA的敲低(shRNA1和2联合应用)。(B) PAK2和PRKACA敲低对SU-DIPGXVII和SU-DIPG36细胞体外培养96小时的生长影响。(C) 与对照细胞相比,PAK2和PRKACA敲低细胞中差异表达蛋白的通路富集分析。(D) PKA和AKT1药物对DMG细胞体外培养72小时的生长影响。
03
METTL13 和 METTL21B 是 DMG 甲基化蛋白质组的关键驱动因子
组蛋白H3K27三甲基化(H3K27me3)的整体降低是H3K27改变的DMG的标志性特征;然而,非组蛋白的甲基化尚未得到全面研究。为了鉴定DMG和正常脑组织中整体蛋白质甲基化情况,特别是非组蛋白的甲基化,他们采用了强阳离子交换(SCX)结合液相色谱-质谱联用(LC-MS)技术。与正常脑组织相比,DMG组织中整体蛋白质甲基化水平显著升高(图1E)。二甲基-K、二甲基-R和三甲基-K在DMG组织中富集最为显著(图1F)。基于甲基化蛋白质组谱,DMG和正常脑组织样本之间存在明显的聚类关系。这种分组似乎具有普遍性,因为在不同 H3 突变状态的 DMG 样本中未观察到明显差异。他们鉴定出 38 个(占捕获的 166 个非组蛋白甲基化蛋白的 23%)显著差异甲基化的蛋白,其中 20 个蛋白在 DMG 组织中与正常脑组织相比呈高甲基化状态,18 个蛋白呈低甲基化状态。这些差异甲基化蛋白与翻译上调(例如 EEF1A1 和 EEF1A2)以及剪接上调(例如 HNRNPA1、HNRNPAB 和 HNRNPA2B1)相关(图 4A)。剪接是一种非诱变机制,可激活胶质瘤中的致癌通路,而该机制的大部分是通过蛋白质甲基化调控的。虽然 GFAP 是甲基化程度最高的蛋白,但翻译延伸蛋白 EEF1A1 是 DMG 中甲基化程度最高的翻译机制蛋白(图 4A)。与正常脑组织相比,DMG 组织中 EEF1A1 的 K79 位点三甲基化和 K55 和 K165 位点二甲基化显著升高(图 4B)。
为了揭示DMG甲基化组的调控因子,他们利用多组学数据,在mRNA和蛋白水平上检测了DMG中非组蛋白甲基转移酶的表达。METTL13、METTL21B和PRMT5的表达显著上调(图4C)。PRMT5 是一种精氨酸甲基转移酶,而 METTL13 和 METTL21B 是 METTL 家族的赖氨酸甲基转移酶,它们能够甲基化 EEF1A1,EEF1A1 是 DMG 中甲基化程度最高的蛋白质之一。已在体外证实了 METTL13 和 METTL21B 对 EEF1A1 蛋白的甲基转移酶活性(图 4D)。有趣的是,在纳入的队列中,EEF1A1 高甲基化与 DMG 的不良预后相关,提示其可能促进更具侵袭性的肿瘤行为(图 4E)。EEF1A1 广泛表达,是细胞中最丰富的蛋白质之一。EEF1A1 是一种必需蛋白,其本身并非可行的治疗靶点,但 METTL13 和 METTL21B 对其的甲基化并非必需修饰,因此可能成为潜在的治疗靶点。重要的是,癌细胞需要高水平的EEF1A1甲基化来满足其翻译需求,因此,METTL13和METTL21B是癌细胞而非正常细胞中的必需蛋白。事实上,CRISPR基因敲除研究表明,METTL13在正常细胞系中并非必需基因,但在骨肉瘤、弥漫性胶质瘤、非霍奇金淋巴瘤、黑色素瘤和脂肪肉瘤细胞系中则更可能是必需基因。METTL13在癌细胞中的扰动基因效应评分显著低于正常细胞系,这表明靶向该甲基转移酶可能对正常组织毒性有限。
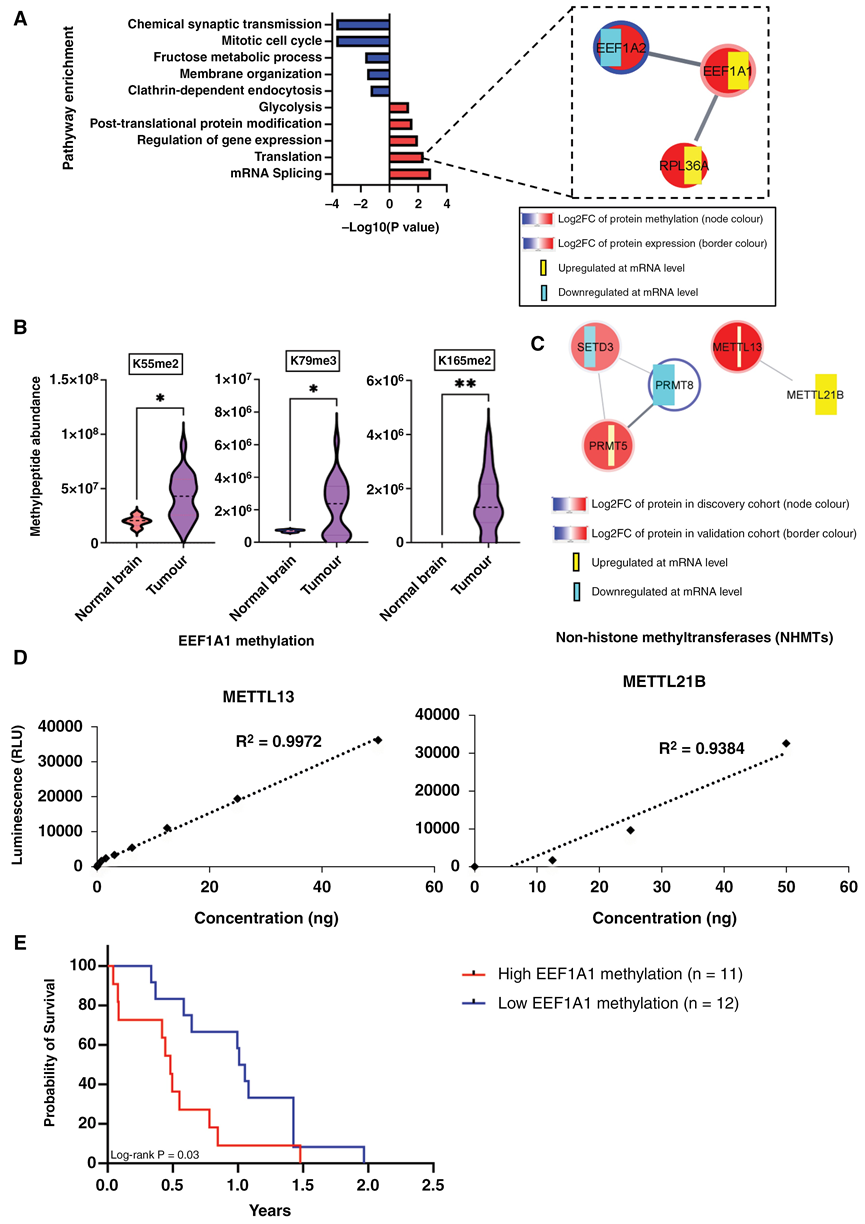
图4. 蛋白质甲基化与DMG中生存率的相关性。
(A) DMG中差异甲基化蛋白的通路富集分析。(B) 非组蛋白EFF1A1在DMG中K55、K79和K165位点高度甲基化。(C) DMG中富集程度最高的5个NHMT(mRNA或蛋白表达)。(D) 体外甲基转移酶活性分析显示METTL13和METTL21B对EEF1A1甲基化的活性。(E) Kaplan-Meier生存曲线显示了EEF1A1甲基化与DMG患者生存率之间的关系,结果显示二者存在显著相关性。
04
METTL13 敲低降低癌蛋白组的表达并降低 DMG 中的细胞活力
在 EEF1A1 和 EEF1A2 这两个亚型中,只有 EEF1A1 在 DMG 中相对于正常脑组织上调。相反,EEF1A2 在正常脑组织中的表达水平极低,在 DMG 中则进一步下调。METTL13 在蛋白水平上高表达,且其底物高度甲基化,表明 METTL13 在 DMG 中具有活性。METTL21B 在 DMG 中 mRNA 水平显著高表达,但在蛋白质组队列中未检测到)。基于质谱的大型蛋白质组学数据中缺失值相当常见,可以通过多重插补方法或靶向蛋白质组学来解决。通过Western blot证实了DMG中EEF1A1、METTL13和METTL21B的表达水平升高。此外,通过从肿瘤蛋白裂解物中免疫沉淀 EEF1A1,然后使用抗泛甲基赖氨酸抗体进行蛋白质印迹分析,验证了 DMG 组织中 EEF1A1 的甲基化。因此,METTL13 和 METTL21B 过表达,并且它们的底物高度甲基化,表明这些非组蛋白甲基转移酶在 DMG 中具有活性,并可能代表一种新的治疗靶点。
由于目前尚无特异性抑制剂可用于研究METTL13和METTL21B抑制对DMG细胞的影响,他们使用了针对每个基因的两种独立的shRNA来降低这些基因的表达。通过Western blot证实了METTL13和METTL21B蛋白水平的敲低,并导致EEF1A1蛋白甲基化水平显著降低,特别是赖氨酸55和赖氨酸165的甲基化(图5B-D)。
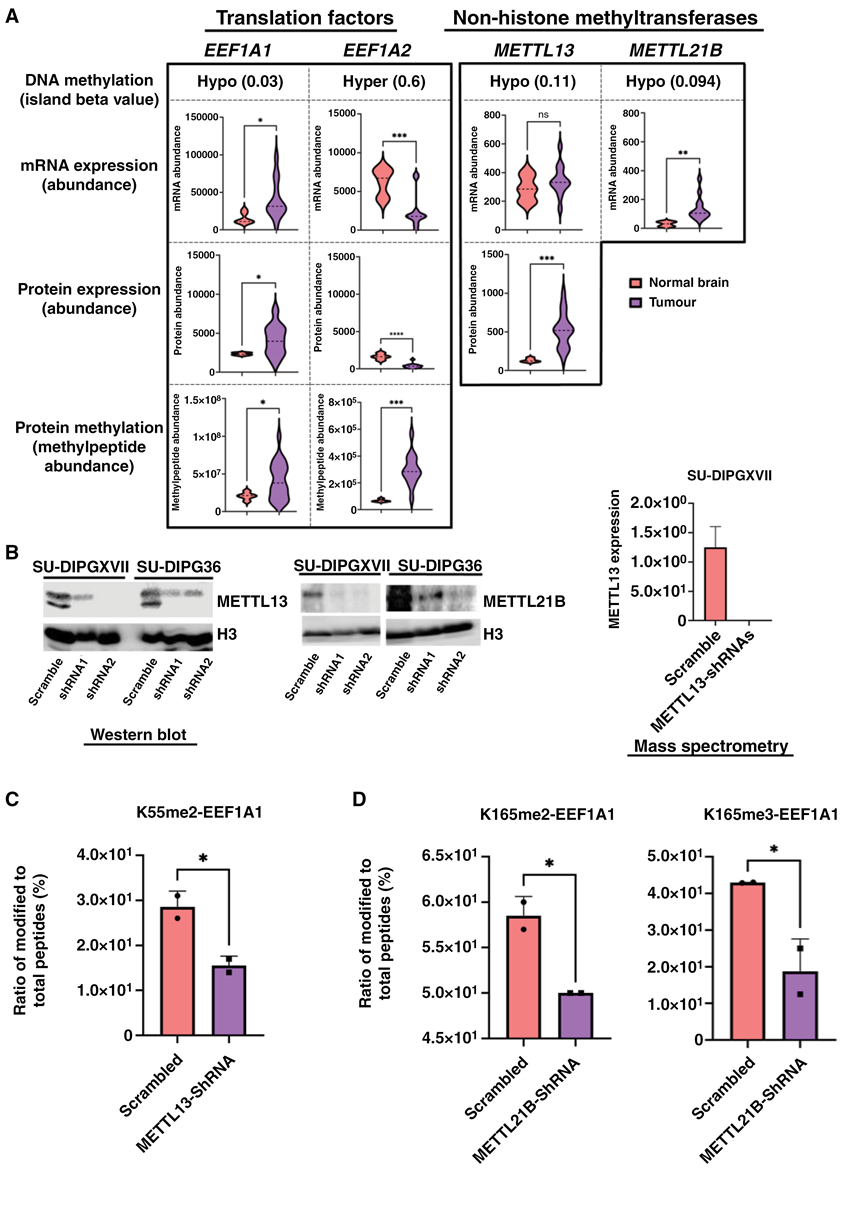
图5. 甲基转移酶候选基因 METTL13 和 METTL21B 在弥漫性胶质瘤 (DMG) 细胞中的作用。
(A) DMG 细胞中 EEF1A1、EEF1A2、METTL13 和 METTL21B 的多组学富集分析。METTL21B 在蛋白质组数据中缺失。(B) 通过蛋白质印迹和质谱分析验证了基于 shRNA 的 METTL13 和 METTL21B 在 DMG 细胞中的敲低效果。(C) 在 SU-DIPG36 细胞中,敲低 METTL13 和 METTL21B 显著降低了 EEF1A1 K55 位点的甲基化水平。(D) 敲低METTL21B显著降低了SU-DIPG36细胞中EEF1A1 K165的甲基化水平。
DMG细胞普遍存在H3K27me3丢失和染色质开放,从而为转录创造了有利环境。由于翻译延伸蛋白EEF1A1高度甲基化,他们推测DMG细胞具有高度翻译活性,甲基化的K55有利于癌蛋白组的形成和细胞生长。他们使用L-2-碲烯基丙氨酸(TePhe)作为非天然氨基酸,通过质谱流式细胞技术直接检测蛋白质合成,无需实验后标记。与NHA细胞相比,DMG细胞系SU-DIPGXVII和SU-DIPG36的蛋白质合成率显著升高(图6A)。METTL13 敲低以及随之而来的 EEF1A1 甲基化减少与 SU-DIPGXVII 和 SU-DIPG36 细胞中蛋白质合成显著降低以及细胞生长减少相关(图 6A-B)。此外,他们在 SU-DIPG25 和正常人星形胶质细胞(NHA,分离自 DMG 患者)中进行了 METTL13 敲低,并观察到敲低不影响 NHA 在体外的生长支持 DMG 对 METTL13 的特异性依赖性。有趣的是,SU-DIPGXVII 和 SU-DIPG36 细胞中蛋白质合成的减少并非普遍现象——相反,METTL13 的敲低和 EEF1A1 K55 位点甲基化的降低导致了一组特定蛋白质的翻译减少。先前的 METTL13 敲除研究表明,METTL13 对赖氨酸(AAA 和 AAG)和组氨酸具有密码子偏好性,即赖氨酸含量高的蛋白质翻译速率较慢,而丙氨酸密码子的翻译速率较快。在 DMG 中,METTL13 敲低以类似的特异性方式影响蛋白质合成,并且伴随着细胞生长减少(图 5D-C)。METTL13 的敲除降低了密码子特异性翻译速率;与对照细胞相比,METTL13 敲低细胞中下调的蛋白质具有较高的赖氨酸和谷氨酸组成(例如,剪接蛋白 SF3A2),而上调的蛋白质具有较高的丙氨酸和亮氨酸组成(例如,丝氨酸蛋白酶 HTRA1)。
总体而言,在DMG中,METTL13通过EEF1A1的甲基化介导其致癌作用,加速富含赖氨酸的蛋白质的翻译,同时抑制富含丙氨酸的蛋白质的翻译,从而形成DMG特有的癌蛋白组。对 METTL13 敲低 DMG 细胞(而非 METTL21B 敲低 DMG 细胞)中差异表达蛋白的质谱分析表明,mRNA 剪接、细胞周期、氨基酸代谢和翻译均有所降低;这些对于维持肿瘤发生至关重要(图 6C)。虽然 METTL21B 敲低对蛋白质合成有普遍影响,但 METTL13 敲低则导致癌蛋白组合成发生改变。这些发现支持以下观点:抑制 METTL13 的甲基转移酶活性不仅会降低蛋白质合成,还会靶向特定的致癌过程(癌蛋白组),从而支持 DMG 细胞的存活。
由于目前尚无针对 METTL13 的药物,他们使用转染了对照 shRNA 或 METTL13 特异性 shRNA 的 SU-DIPG36 细胞,在体内评估了 METTL13 敲低对 DMG 异种移植模型的影响。在进行体内实验之前,通过Western blot证实了 SU-DIPG36 细胞中 METTL13 的敲低,蛋白质水平的敲低率约为 60%。与注射对照 DMG 细胞的小鼠相比,注射 METTL13 敲低 DMG 细胞的小鼠生存期显著延长,尽管 METTL13 敲低程度相对较低(图 6D)。这项临床前验证凸显了靶向 METTL13 治疗 DMG 患者的潜力。本研究全面分析了DMG的蛋白质组、磷酸化蛋白质组和甲基化蛋白质组,从而构建了激酶和甲基转移酶活性及其对DMG影响的整合模型(图6E)。
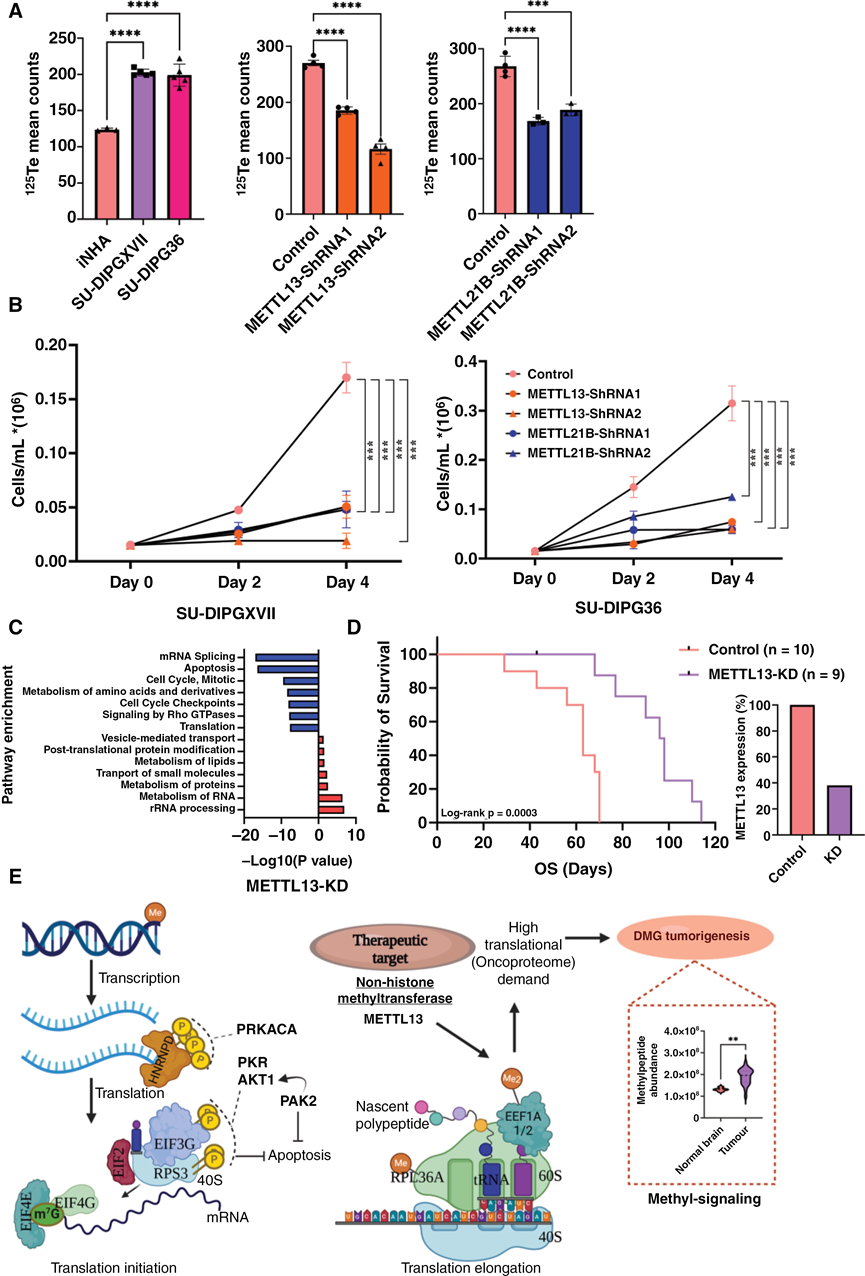
图6. 敲低 METTL13 和 METTL21B 可降低体外整体蛋白质合成和细胞生长。
(A) 敲低 METTL13 和 METTL21B 显著降低了 SU-DIPG36 细胞的整体蛋白质合成。DMG 细胞的蛋白质合成显著高于对照 iNHA 细胞。(B) 敲低 METTL13 和 METTL21B 对体外 DMG 细胞生长的影响。(C) 与对照细胞相比,METTL13 敲低细胞中差异表达蛋白的通路富集分析。(D) 在体内 DMG 异种移植模型中,敲低 METTL13 可提高小鼠的生存率。对照组和 METTL13-KD 小鼠的中位生存期分别为 63 天和 97 天。本实验使用了 METTL3-ShRNA2。(E) 激酶和 METTL13 底物及其对翻译机制和 DMG 肿瘤发生的影响的整合模型。
+ + + + + + + + + + +
结 论
本研究利用液相色谱-质谱联用技术(LC-MS)对55例DMG患者样本进行了全面的蛋白质组学分析,并结合DNA甲基化和DNA/RNA测序。此外,还对其中30例患者样本进行了翻译后修饰谱分析(磷酸化蛋白质组和甲基化蛋白质组)。DMG 表现出高度的整体蛋白质甲基化,其中翻译机制蛋白和参与细胞凋亡调控的因子显著富集。令人惊讶的是,关键激酶的靶蛋白高度磷酸化,但 DMG 中的整体蛋白质磷酸化水平低于正常脑组织。非组蛋白甲基转移酶 METTL13 和 METTL21B,以及蛋白激酶 PAK2、PRKACA 和 AKT1,分别鉴定为 DMG 甲基化蛋白质组和磷酸化蛋白质组变化的关键参与者。METTL13 敲低导致 EEF1A1 蛋白甲基化水平降低,癌蛋白合成发生改变,并在体外和体内均抑制 DMG 细胞的生长。
+ + + + +



