

 English
English文献解读|Nat Genet(29.0):人类视网膜转录组和染色质可及性的单细胞图谱
✦ +
+
论文ID
原名:Single-cell atlas of the transcriptome and chromatin accessibility in the human retina
译名:人类视网膜转录组和染色质可及性的单细胞图谱
期刊:Nature Genetics
影响因子:29.0
发表时间:2026.01.23
DOI号:10.1038/s41588-025-02454-1
背 景
高通量单细胞转录组学技术的出现极大地促进了对细胞多样性的探索。这些技术能够构建单细胞分辨率的综合图谱,这对于研究细胞功能和疾病机制至关重要。人类细胞图谱(HCA)项目协调了多项合作计划,旨在对整个人体内的细胞类型进行编目,迄今为止已发布了组织特异性图谱。在HCA内部,眼生物学网络致力于构建人眼细胞图谱。近期研究已构建了人眼前段和后段的图谱。其他研究则构建了包括小鼠、鸡、猕猴和人类在内的多种物种的视网膜图谱。本文旨在通过纳入更多供体、细胞和方法,扩充先前的数据集,从而构建一个全面的人类视网膜细胞图谱。
实验设计
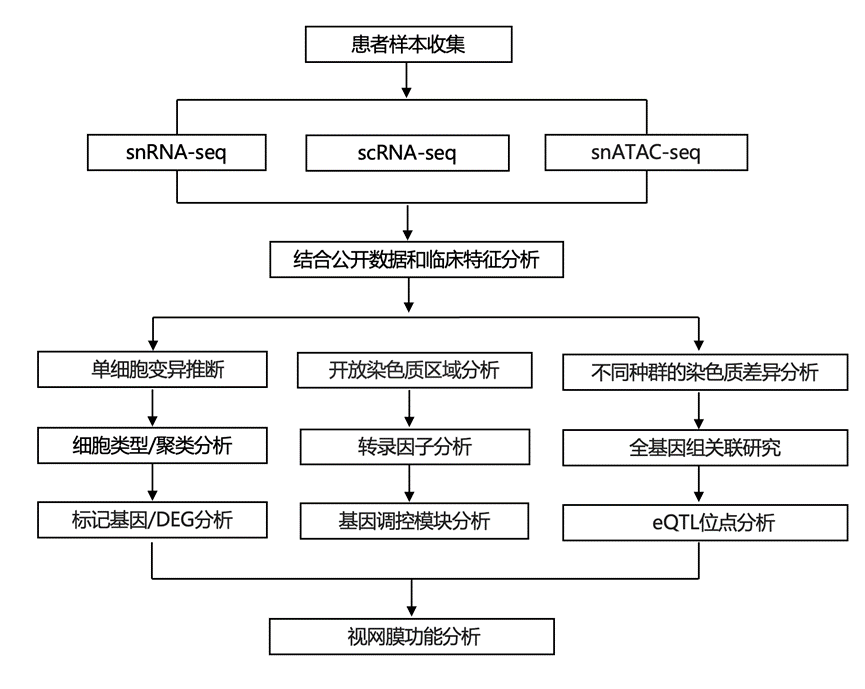
结 果
01

人类视网膜单细胞图谱
为了获得人类视网膜细胞类型的全面图谱,研究团队将八个公开数据集与新生成的未发表数据整合在一起(图1a-b)。整合后的数据集包含来自122位供体共280个样本的3443077个单核和单细胞转录组。他们使用一种高级标签预测方法对各个样本中的主要视网膜细胞类型进行了注释,这些细胞类型包括星形胶质细胞、无长突细胞(AC)、双极细胞(BC)、视锥细胞、水平细胞(HC)、胶质细胞(MG)、小胶质细胞、视网膜神经节细胞(RGC)、视网膜色素上皮细胞(RPE)和视杆细胞。为了容纳大量的细胞,我们对所有细胞进行了数据整合,以便对BC、AC和RGC进行谱系特异性注释,因为它们具有复杂的细胞类型。主要细胞类别分布一致,但部分供体样本中AC和RGC的富集情况除外。这些供体样本经过细胞富集处理,提高了高度异质性细胞类别(AC、BC和RGC)的比例,从而便于对稀有细胞类型进行注释。
他们使用单细胞变异推断(scVI)技术分析,基于先前的基准研究,整合了整个数据集,并使用均匀流形近似和投影 (UMAP) 将所有细胞嵌入二维空间。他们比较了单细胞转录组分析(scRNA-seq)数据和单细胞核转录组分析(snRNA-seq)数据在此UMAP中的分布,发现它们的转录组特征存在显著差异,因此无法使用 scVI 将 snRNA-seq 和 scRNA-seq 数据进行比对。为了评估这些差异,他们使用伪批量分析对280个样本的表达进行平均分析,并证实 snRNA-seq 和scRNA-seq产生了不同的转录组数据,这与之前的报道一致。因此,他们为snRNA-seq(图1c)和scRNA-seq分别创建了两个独立的参考序列。这两个参考序列均通过经典标记基因的表达进行了验证(图1d)。
为了进一步比较snRNA-seq和scRNA-seq,他们分析并比较了中央凹、黄斑和周边区域的细胞比例。最显著的差异是,在中央凹和黄斑区域,scRNA-seq数据集中的MG细胞比例高于snRNA-seq数据集。两种技术的细胞簇可以很容易地进行比对,因为它们具有相似的主要细胞类别的转录组特征(图1e)。然而,两种技术之间检测到了许多差异表达基因(DEG)。总共在snRNA-seq和scRNA-seq数据集的所有细胞类型中鉴定出1815个和5473个过表达基因(图1f)。这些基因表现出不同的但生物学上相关的富集基因本体生物学过程。
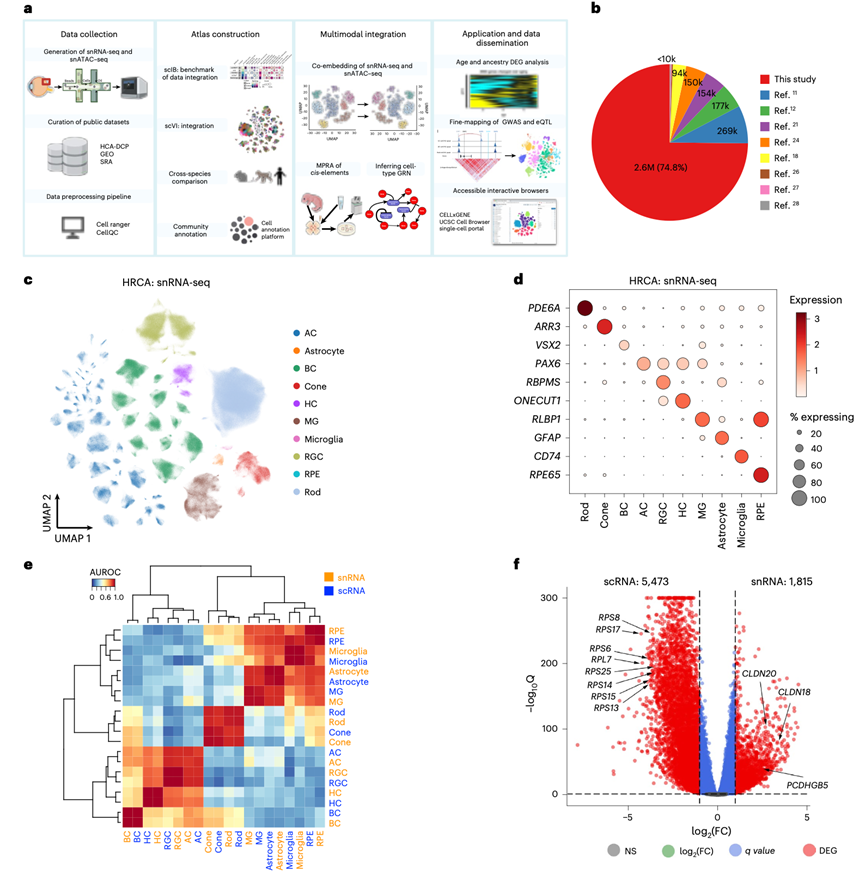
图1. 人类视网膜单细胞图谱概览。
(a) 实验概览。(b) 收集的视网膜数据集包括内部新生成的数据集和八个公开可用的数据集。(c) snRNA-seq 数据集图谱以 UMAP 图的形式可视化,分辨率为主要类别,细胞根据其主要类别着色。(d) 点图展示了 snRNA-seq 数据中主要细胞类别标记基因表达水平的分布。(e) snRNA-seq(珊瑚色)和 scRNA-seq(蓝色)之间主要类别的细胞类型相似性。(f) snRNA-seq数据集(右)和scRNA-seq数据集(左)中过表达基因的火山图。(g) (h) (i) (j) (k) (l) (m) (n)
02
BC 类型的跨物种分析
本图谱中包含的超过69万个双极单核细胞根据标记基因可鉴定为14种细胞类型(图2a)。值得注意的是,巨型双极细胞(GB)和蓝色双极细胞(BB)形成了两个不同的聚类,这可能是由于与之前的报道相比,细胞数量显著增加所致(图2b)。为了便于注释,他们进行了跨物种分析,利用单细胞转录组和蛋白质序列嵌入,将人类双极细胞聚类与小鼠和猕猴的双极细胞类型进行比对(图2c-d)。在三个物种之间观察到高度一致的一对一映射,这与之前的报道一致。基于共嵌入分析,映射到小鼠细胞类型BC9的人类基因聚类注释为BB细胞,因为已有报道BC9细胞特异性地与S锥细胞(在人类和猕猴中也称为“蓝色”锥细胞)接触;而映射到BC8细胞的人类基因聚类则注释为GB细胞。虽然GB细胞和BB细胞之间存在相似性,但GB细胞中高表达基因有327个,而BB细胞中高表达基因有941个。其中,AGBL1和SORCS3分别对GB细胞和BB细胞表现出高度特异性(图2a)。与此一致,在scRNA-seq数据集中也观察到了14个BC对应的基因聚类。此外,GB和BB中的差异表达基因(DEG),包括AGBL1和SORCS3,也通过scRNA-seq得到验证,在GB中重叠率为58%,在BB中重叠率为12%(图2a-e)。
在小鼠中,已鉴定出以下四种BC5类型:BC5A、BC5B、BC5C和BC5D。然而,这些密切相关的BC类型与灵长类动物中的BC之间的关联尚未完全阐明。此前,只有小鼠BC5A被证实与猕猴DB4存在可靠的对应关系。两种人类BC类型DB4a和DB4b与小鼠BC5A和猕猴DB4密切相关,而小鼠BC5B和BC5C似乎与人类和猕猴DB5最为相似(图2f)。相比之下,小鼠BC5D似乎是一个特例,在灵长类动物中缺乏与其密切相关的BC类型。为了区分这些BC类型,他们鉴定了一组55个基因标记,这些标记表现出良好的性能。
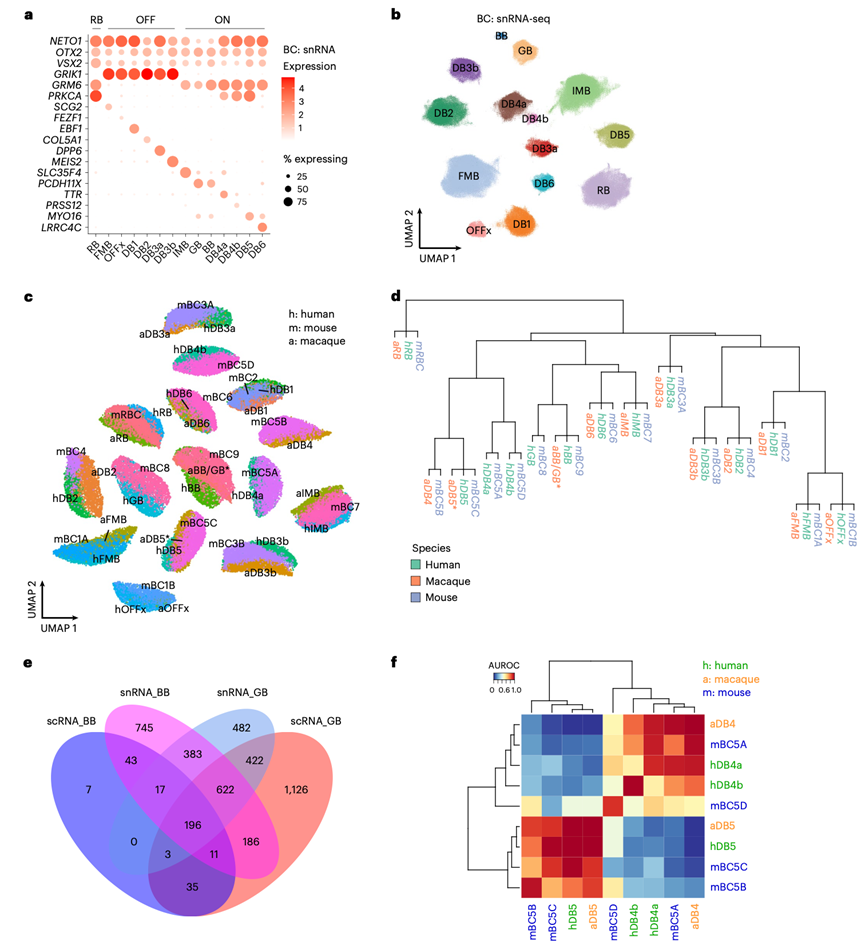
图2. BC 类型在人类、猕猴和小鼠视网膜中高度保守。
(a) BC类型标记基因的分布。(b) 人类BC类型的UMAP可视化。(c) 人类、小鼠和猕猴BC的共嵌入。(d) 小鼠BC类型的层次聚类。(e) 利用snRNA-seq和scRNA-seq数据集,分析了人类GB和BB中排名靠前的基因的重叠情况。(f) 小鼠BC5A、BC5B、BC5C和BC5D细胞类型与人类和猕猴中已定位细胞类型的相似性。
03
无长突细胞和视网膜神经节细胞分析
在超过571000个AC细胞核中,共鉴定出89个AC聚类(图3a),几乎是先前报道的AC类型数量的两倍。两种AC泛标记物PAX6和TFAP2B在这些聚类中高表达。利用GABA能AC(GABA合成酶GAD1和GAD2)和甘氨酸能AC(甘氨酸转运蛋白SLC6A9)的标记物,他们鉴定出64个GABA能AC聚类(约占AC总数的67%)和15个甘氨酸能AC聚类(约占AC总数的23%)。有10个聚类同时表达这两种标记物,并归类为“两种”AC类型。基于先前已鉴定的其他标记物,对已知的14种腺泡细胞类型进行了注释(图3a)。例如,基于CHAT和MEGF10的表达,HAC10注释为ON型AC细胞,而基于CHAT和TENM3的表达,HAC31注释为OFF型AC细胞。他们鉴定了一组能够区分所有89种AC细胞类型的基因标记物。为了进一步注释AC细胞类型,我们采用交叉比对方法,将鉴定的AC细胞聚类与外部数据集和其他物种(包括猕猴和小鼠)进行比对。观察到高度一致性——88%的scRNA-seq聚类映射到该数据集,94%(34个中的32个)的猕猴AC类型成功映射到人类数据集。相比之下,仅有81%(63个中的51个)的小鼠AC类型映射到人类,其中包括4个非GABA能非甘氨酸能(nGnG)小鼠类型映射到人类聚类。17个人类聚类与先前注释的类型缺乏明确的对应关系。这些聚类似乎很罕见,其中最丰富的聚类仅占AC总数的0.18%(677个细胞核)。
他们在图谱中超过372,000个RGC核中鉴定出24个RGC聚类(图3b)。利用先前已鉴定的猕猴标记物,他们注释了六种类型:TBR1标记的OFF型小RGC(MG_OFF)、TPBG标记的ON型小RGC(MG_ON)、FABP4标记的OFF型伞状RGC(PG_OFF)、CHRNA2标记的ON型伞状RGC(PG_ON)、OPN4标记的内在光敏RGC(ipRGC)以及BNC2标记的ON型方向选择性RGC(DS_ON)。与之前的报道一致,人类RGC类型的分布高度偏斜,其中小型RGC占88.1%。伞状视网膜神经节细胞(Parasol RGC)占比4.1%,与之前的研究相比,其代表性不足,这可能是由于实验富集所致。人类、猕猴和小鼠之间的跨物种比较表明,RGC类型差异很大(图3c)。由于灵长类动物的RGC类型(约18种)与小鼠的RGC(45种分子不同的类型)相比,多样性显著降低,因此难以在人类和小鼠之间进行聚类分析。最后,他们使用二元分类方法和NS-Forest鉴定了一组RGC聚类的标记(图3d)。
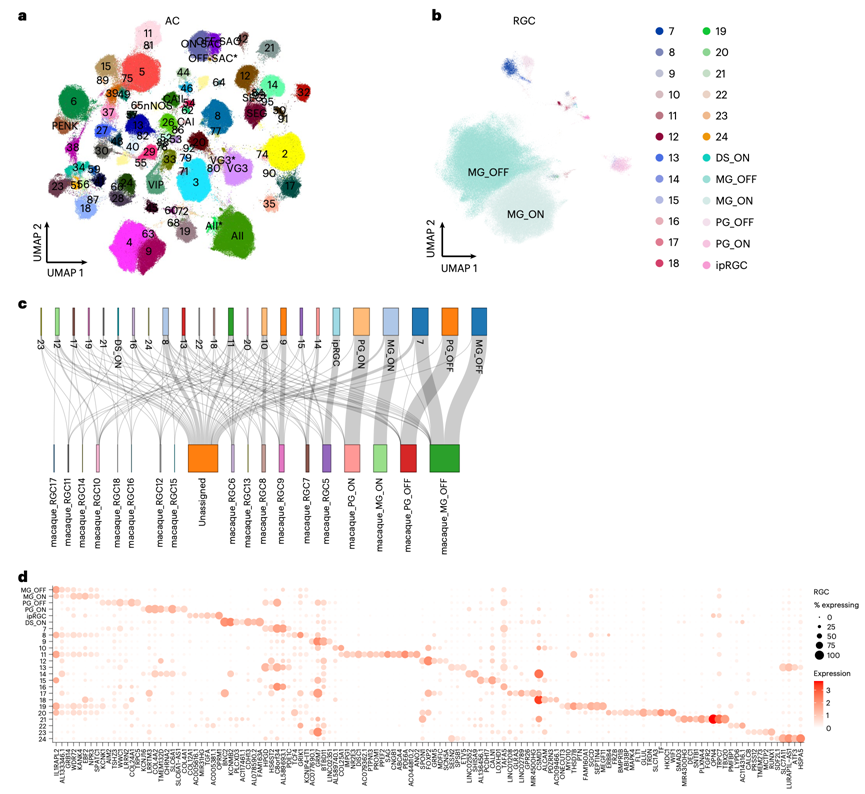
图3. AC 和 RGC 类型在人类、猕猴和小鼠视网膜中表现出越来越大的差异。
(a) UMAP可视化图展示了已识别的89个AC聚类。(b) UMAP可视化图展示了RGC类型。(c) 桑基图展示了人类(左列)和猕猴(右列)的RGC类型比对情况。(d) RGC类型预测标记的散点图。
04
HRCA:染色质可及性图谱
为了解析视网膜细胞类型的基因调控程序,他们收集了经质量控制后的477180个细胞核的snATAC-seq数据,其中包括来自公共数据集的309608个细胞核和来自未发表数据的167572个细胞核。这些细胞核注释为六种神经元和三种胶质细胞(图4a-b)。全基因组基因表达(包括经典标记基因)与所有细胞类型的局部染色质可及性和推断的基因活性高度相关(图4c)。
基于该数据集,他们鉴定了700146个开放染色质区域(OCR),每个细胞类别包含135,675至218,488个OCR(图4d)。为了评估这些OCR的质量,他们将其与单细胞核转座酶可及染色质测序分析(snATAC-seq)检测到的OCR进行了比较。snATAC-seq技术捕获了批量ATAC-seq检测到的大部分(77.4%)OCR,并鉴定出更多细胞类别特异性OCR,使OCR总数增加了三倍。许多OCR在多个细胞类别中共存,但6.1%至20.9%的OCR表现出不同的可及性,这取决于细胞类别,这表明它们可能在细胞类别特异性基因调控中发挥作用;他们将这些 OCR 称为差异可及区域(DAR)。 与已验证的沉默子和非活性元件相比,它们富含视杆细胞转录因子 (TF) 的结合和活性增强子的表观遗传修饰(图4g)。此外,他们利用SCENIC整合了snRNA-seq和snATAC-seq数据,鉴定了主要细胞类型的转录因子。许多已鉴定的TF已被先前的研究证实与视网膜特定细胞类型的特化有关。
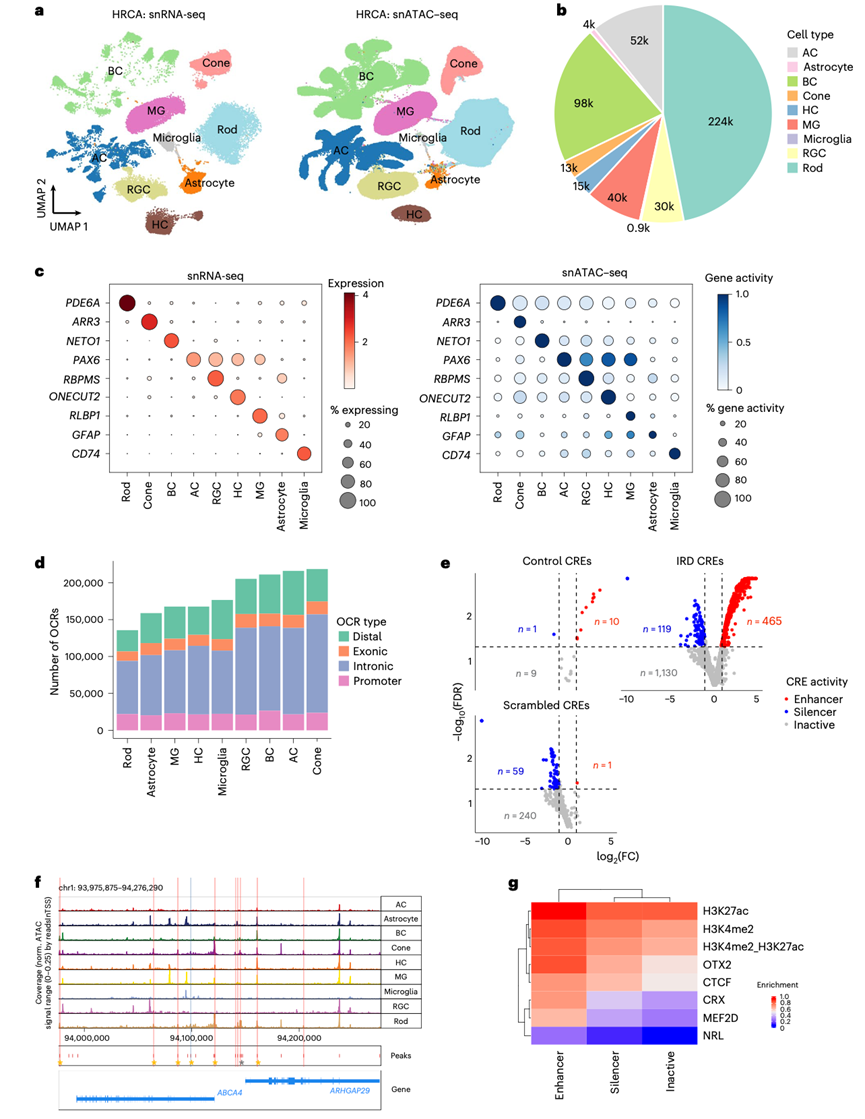
图4. 人类视网膜高分辨率染色质可及性细胞图谱。
(a) snRNA-seq 和 snATAC-seq 共嵌入细胞的 UMAP 图,显示细胞聚类为主要的视网膜细胞类型。(b) 饼图显示主要视网膜细胞类型的细胞比例分布。(c) 散点图显示 snRNA-seq 检测的标记基因表达和 snATAC-seq 获得的相应细胞类型的标记基因活性评分。(d) 柱状图显示每个主要细胞类型中鉴定的 OCR 数量。(e) 火山图显示CRE活性。(f) 基因组轨道显示了ABCA4基因座周围的假定CRE。(g) 热图显示了基于已发表的ChIP-seq数据集。
05
人类 BC 类型的调节子
他们使用GLUE方法整合snRNA-seq 数据,进一步细化了 snATAC-seq 类别中的细胞类型注释,鉴定了14种BC类型,与 snRNA-seq 注释的 14 种类型相符(图5a)。其中两种 snATAC-seq 类型,GB和BB,与 snRNA-seq 注释结果一致。与此相符的是,GB 和 BB 的标记基因UTRN和SORCS3(由 snRNA-seq 鉴定)分别表现出细胞类型特异性的染色质可及性(图5b-c)。类似地,他们也对其他异质性神经元类别(AC、HC、视锥细胞和 RGC)和非神经元类别(MG、星形胶质细胞和小胶质细胞)中的细胞类型进行了注释。
与BB和GB类似,其他细胞类型也受多种TF的调控。他们观察到TF的协同作用,这体现在这些TF靶区域和靶基因的重叠上(图5d)。例如,NFIA的部分靶区域和靶基因与MEIS2、NEUROG1和ZNF385D的靶区域和靶基因重叠,这与它们靶区域的高可及性和在DB3b细胞中的高TF表达相一致(图5d)。NFIA的靶区域和靶基因也与NFIX和POU6F2的靶区域和靶基因重叠,这与它们靶区域的高可及性和在DB4b细胞中的高TF表达相一致(图5d)。这些发现表明,同一个转录因子可以在不同的细胞类型中与不同的转录因子协同作用。
为了进一步鉴定驱动BC类型的调控子组合,他们基于调控子的活性计算了它们之间的相关性,最终得到14个调控子模块。大多数模块在特定的BC亚群中表现出更高的曲线下面积(AUC)值,尤其是在那些转录组谱更相似的亚群中(图5e)。总之,这些观察结果表明,每种细胞类型都由独特的TF组合编码定义,该编码是通过精确调控TF表达及其靶区域的染色质状态而建立的。
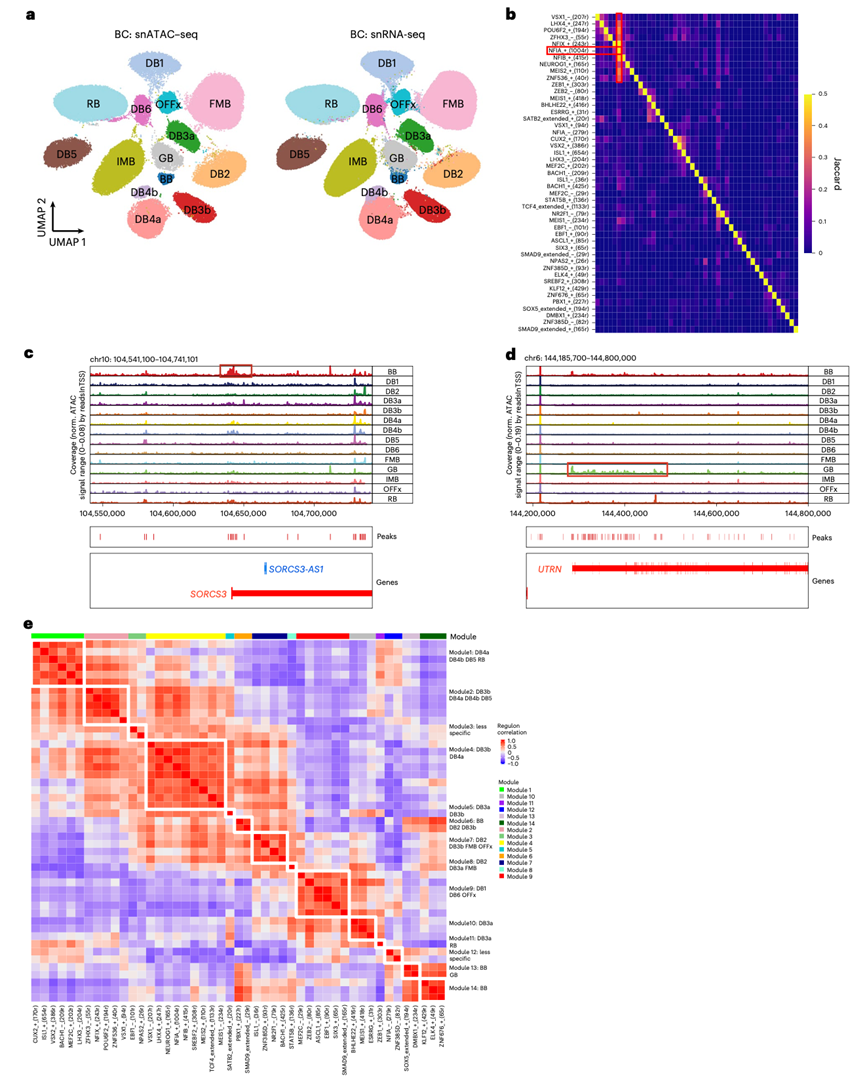
图5. 人类 BC 类型的调节子。
(a) snRNA-seq 和 snATAC-seq 共嵌入细胞的 UMAP 图,显示细胞聚类为 BC 类型。(b) Jaccard 热图,显示已鉴定转录因子靶区域的交集。(c) SORCS3的基因组轨迹,显示SORCS3的启动子在 BB 中高度开放。(d) UTRN的基因组轨迹,显示UTRN的局部染色质在 GB 中高度开放。(e) 热图显示基于靶区域可及性 AUC 值鉴定的调控子的相关性。
06
与年龄和组织区域相关的差异基因表达
视网膜功能和疾病风险的差异与年龄相关。他们分析了来自91名供体(59名男性和32名女性,年龄3至91岁)的211个样本在衰老过程中的转录组动态变化。他们使用线性混合效应模型(LMM)在不同细胞类型中鉴定出131至2820个年龄依赖性基因,该模型考虑了混杂因素(批次效应、祖先背景、性别、组织区域),并使用多元自适应收缩(MASH)方法比较了不同细胞类型之间的效应(图6a)。年龄相关的DEG在不同细胞类型中均表现出正相关性,且神经元类型之间的相关性强于神经元类型与非神经元类型之间的相关性(图6b)。随着年龄增长而上调的基因富集于胞吐作用、核苷酸磷酸化、微管束形成、纤毛或鞭毛依赖性细胞运动以及轴丝组装的调控(图6c)。相反,下调的基因富集于端粒维持、核糖核蛋白复合物生物合成、RNA剪接、RNA代谢过程、蛋白酶体蛋白分解代谢过程、胞质翻译、染色质重塑以及WNT介导的细胞间信号传导(图6d)。这些结果表明,随着年龄增长,基因调控稳态发生改变,其特征是RNA加工、代谢和生物合成活性降低。
他们还观察到每种细胞类型中中央视网膜和周边视网膜的转录组差异。成对比较显示,每种细胞类型中,中央凹/旁中央凹区域与周边视网膜区域之间存在374至3388个差异表达基因(DEG),黄斑区域与周边区域之间存在110至1515个DEG(图6e-f)。虽然许多区域依赖性差异表达基因具有细胞类型特异性,但一些排名靠前的差异表达基因,包括一些TF(如ZIC1,它对视网膜模式形成和细胞分化至关重要),在不同细胞类型中是共存的,这表明存在共同的和依赖于特定环境的调控程序。与中央凹/旁中央凹与周边区域相比,区分黄斑和周边区域的差异表达基因(DEG)数量较少,且超过一半的黄斑-周边区域差异表达基因与中央凹/旁中央凹-周边区域差异表达基因重叠。这表明从中央凹到视网膜周边存在转录组差异的梯度。区域相关的差异表达基因富集于细胞类型特异性和共有的生物学过程。与周边视网膜相比,中央凹/旁中央凹上调的基因富集于核糖体生物合成、氧化磷酸化、前体代谢物和能量的生成、ATP合成偶联电子传递和有氧呼吸,表明其代谢和生物合成活性更高(图6g)。相比之下,外周上调的基因富集于 G 蛋白偶联受体信号通路、纤毛运动、轴丝组装和轴突发育,表明外周视网膜中存在更复杂的细胞相互作用和连接(图6h)。

图6.与年龄和组织区域相关的差异表达基因。
(a) 热图展示了利用线性混合模型(LMM)鉴定的视网膜细胞类型中,衰老过程中DEG的动态变化。(b) 热图展示了利用多因素分析(MASH)确定的细胞类型中,衰老过程中差异基因表达效应大小(右上三角形)和效应方向(左下三角形)的相关性。(c-d) 富集分析。(e) 火山图展示了视杆细胞中中央凹/旁中央凹和周边视网膜之间的DEG。(f) 火山图展示了视神经球状细胞(MG)中中央凹/旁中央凹和周边视网膜之间的DEG。(g-h) 富集分析。
07
转录组和染色质景观中的祖先差异
本研究的snRNA-seq数据集包含来自不同种族背景的样本,从而能够进行与种族相关的转录组和染色质差异的细胞类型水平分析。他们平均在每个细胞类型中鉴定出2847个与美洲混血相关的DEG、2019个与欧洲血统相关的DEG和1742个与非洲血统相关的DEG。神经元细胞类型之间的差异表达效应方向和大小高度相关,但神经元细胞类型与非神经元细胞类型之间的相关性较低。非洲血统比例较高的基因下调,富集于免疫过程,包括对干扰素-γ的反应、炎症反应的调节、体液免疫反应和补体激活(例如,CFI、C3)图7a-b),而欧洲血统比例较高的基因上调,富集于免疫效应过程、体液免疫反应和慢性炎症反应通路(图7c-d)。
利用snATAC-seq技术,他们鉴定了不同种族间全基因组差异可及的开放染色质区域(OCR),称为祖先DAR。他们为每个种族和细胞类型鉴定了多达887个祖先DAR,其中混血美洲人群的祖先DAR数量更多,与DEG模式相似。与全基因组OCR相比,许多祖先DAR位于启动子区域。为了验证这些祖先DAR,他们检测了与祖先DAR启动子相关的基因表达,并观察到其与不同祖先人群启动子可及性存在显著相关性(图7e)。例如,在视锥细胞中,ZNF467的启动子及其CRE在非洲裔个体中均表现出更高的可及性,这与ZNF467表达增加相一致(图7f-g)。
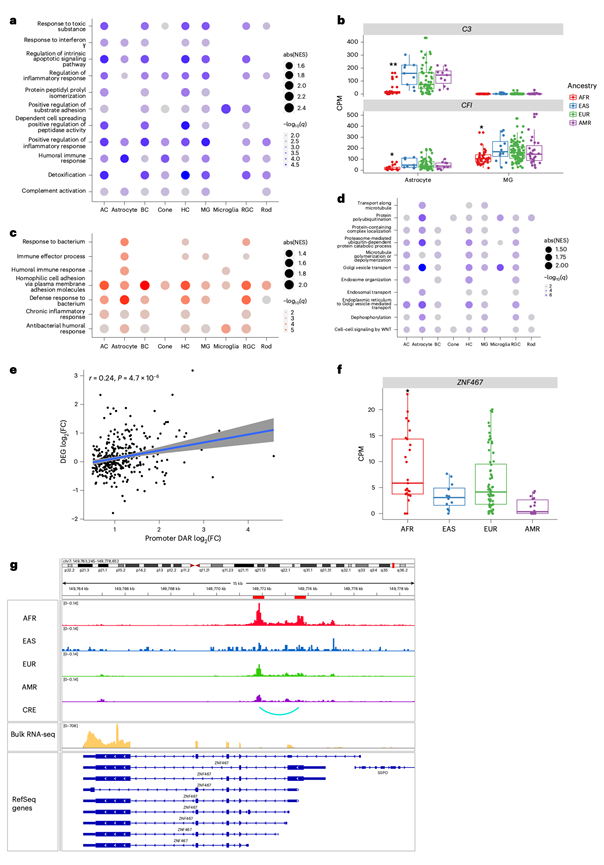
图7. 与祖先背景相关的转录组和染色质可及性差异。
(a, c, d) 富集分析。(b) 不同祖源背景的样本中,星形胶质细胞和髓质细胞中C3和CFI的表达情况。(e) 散点图显示了祖先DAR启动子区域染色质可及性改变与其相关基因表达变化之间的相关性。(f) 箱线图显示了不同祖先背景样本中视锥细胞中ZNF467基因的标准化表达水平。(g) IGV 视网膜 snATAC-seq 和批量 RNA-seq 数据的快照显示祖先 DAR 调控ZNF467,包括启动子区域和与ZNF467相关的 CRE。
08
利用HRCA研究GWAS和eQTL位点
HRCA能够对全基因组关联研究(GWAS)相关的候选致病变异、基因和受影响的细胞类型进行多模态优先级排序。他们鉴定出富集GWAS性状的细胞类别,与snRNA-seq和snATAC-seq的结果一致(图8a)。年龄相关性黄斑变性(AMD)的基因在RPE和小胶质细胞中富集。外节厚度、内节厚度和外核层厚度的基因位点在视杆细胞、视锥细胞、RPE和MG中富集。开角型青光眼相关的性状在MG和星形胶质细胞中富集。屈光不正和近视的基因位点在大多数视网膜细胞类别中均有富集。不同祖先群体的富集模式一致(图8a),表明存在广泛共有的致病变异。
为了确定候选变异、靶基因和细胞类型的优先级,他们对以下七种视网膜性状的基因位点进行了精细定位:外节厚度、内节厚度、外核层、原发性开角型青光眼(POAG)、AMD、屈光不正/近视和糖尿病视网膜病变。利用 GWAS 汇总统计数据和“单效应总和”统计模型(susieR),他们在 95% 可信区间内鉴定了18805 个变异。整合基因组注释信息(包括单细胞多组学数据集),他们将这些变异精细化到功能区域,包括OCR(3819个)、DAR(1012个)、顺式调控元件区(CRE,723个)、启动子(586个)、非翻译区(UTR,312个)和外显子(235个)(图8b)。许多包含精细定位变异的CRE(45.3%)、DAR(33.2%)和OCR(23.5%)均显示出活性增强子的表观遗传修饰(图8c)。结合人类成年视网膜 Hi-C 数据和批量 eQTL 数据,进一步揭示了可能影响基因调控的变异,包括 679 个通过 Hi-C 环与其他区域相连的变异,207 个通过 Hi-C 环与启动子区域相连的变异,31 个与启动子相连且与我们在单细胞数据集中鉴定的 CRE-基因对重叠的变异,19 个与启动子相连且与视网膜 eQTL-eGene 对重叠的变异,以及 17 个与启动子相连且同时与CRE-基因对和 eQTL-eGene 对重叠的变异(图8d)。对大量视网膜 eQTL 应用类似的方法,结果显示出类似的模式,其中 GWAS 变异体在 CRE 中更富集,而 eQTL 变异体在启动子区域更富集(图8e-g)。
这些精细定位的变异位点为GWAS位点的调控机制提供了候选基因和假设。例如,一个POAG变异位点(rs3777588)精细定位至CLIC5 CRE(图8h),该区域在MG中特异性开放,并与CLIC5表达相关。与此一致的是,CLIC5在MG中高表达。GWAS信号通过该变异位点与视网膜CLIC5 eQTL共定位。预测该变异位点可增强HSF1结合,从而可能增强MG中CLIC5的表达,这与eQTL分析中该变异位点的效应方向一致。
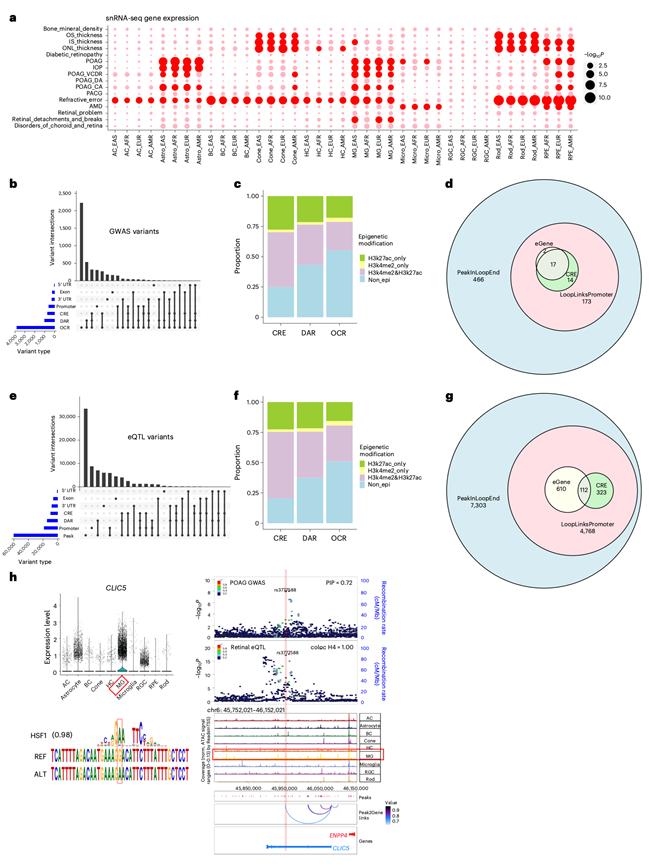
图8. 利用多组学数据研究 GWAS 和 eQTL 位点。
(a) 基于祖先基因表达,利用 MAGMA.Celltyping 对 GWAS 位点进行细胞类型富集分析。(b) 对位于不同基因组区域的精细定位 GWAS 变异进行分类。(c) 包含精细定位 GWAS 变异的 OCR 上表观遗传标记的分布。(d) 利用snATAC-seq和Hi-C-seq数据对精细定位的GWAS变异进行注释。(e) 对位于不同基因组区域的精细定位eQTL变异进行分类。(f) 包含精细定位eQTL变异的OCR上表观遗传标记的分布。(g) 利用snATAC-seq和Hi-C-seq数据对精细定位eQTL变异进行注释。(h) CLIC5区域中精细定位位点的可视化。
+ + + + + + + + + + +
结 论
本研究介绍了一个整合的双模态参考图谱,该图谱涵盖了哺乳动物中枢神经系统最易于获取的部分——视网膜。本研究收集了来自125位具有不同祖源背景的供体的约390万个细胞,其中包括8项已发表的研究和270万个未发表的数据点,从而构建了一个包含130多种细胞类型的综合性人类视网膜细胞图谱。本研究对每个细胞簇进行了注释,鉴定了标记基因,并表征了顺式调控元件和基因调控网络。本研究示了不同细胞类型在转录组、染色质和基因调控网络方面的差异。本研究还模拟了基因表达和染色质可及性随年龄、祖源和组织区域的变化。该整合图谱也增强了GWAS和eQTL变异的精细定位。该多模态、多供体、多实验室的人类视网膜细胞图谱可通过交互式浏览器访问,有助于更好地了解视网膜功能和病理。
+ + + + +



