

 English
English文献解读|Nat Microbiol(20.5):人类阴道微生物组的 33804 个参考基因组的跨界集合
✦ +
+
论文ID
原名:A multi-kingdom collection of 33,804 reference genomes for the human vaginal microbiome
译名:人类阴道微生物组的 33804 个参考基因组的跨界集合
期刊:Nature Microbiology
影响因子:20.5
发表时间:2024.06.21
DOI号:10.1038/s41564-024-01751-5
背 景
人类阴道内藏有各种微生物——细菌、病毒和真菌——对女性健康有着深远的影响。跨多个界别的阴道微生物组的基因组水平分析仍然有限。
近年来,在应用宏基因组组装基因组 (MAG) 分箱方法从培养和未培养的微生物中获取大量基因组信息方面取得了重大进展。这种方法已证明可有效探索人体各个部位的微生物组,包括肠道、口腔和皮肤。多项研究全面收集了超过 150000 个代表人类微生物群的 MAG。然而,该数据集仅包含来自阴道的151 个基因组。因此,迫切需要开展基于宏基因组的研究,旨在建立专门针对阴道微生物群的扩展参考基因组数据库。
实验设计
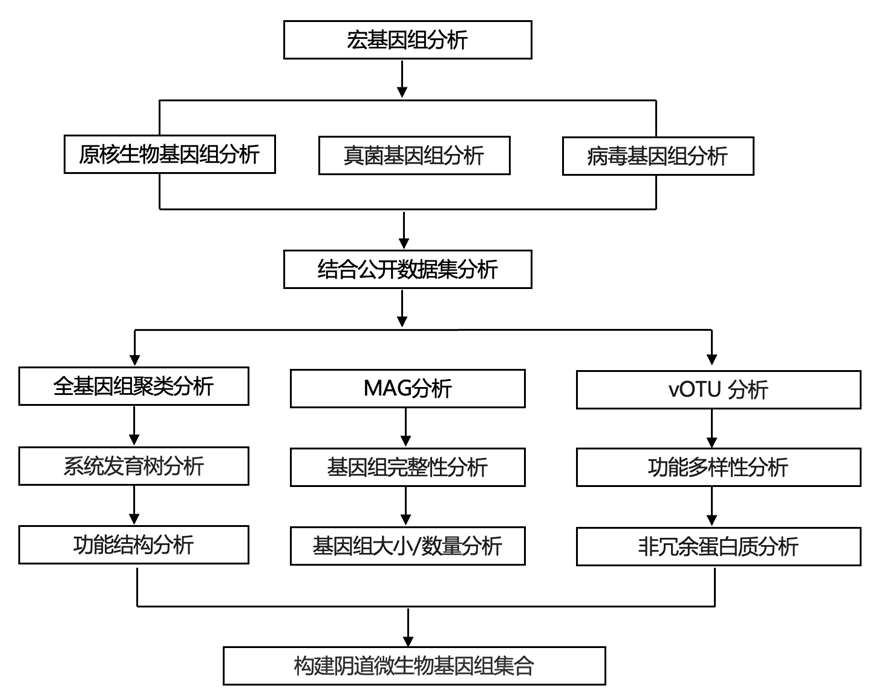
结 果
01

构建阴道微生物基因组集合 (VMGC)
研究团队收集了总共 4472 个可公开获取的人类阴道宏基因组样本,涵盖美国、法国、中国和其他 11 个跨国界国家的 32 项研究(图1a)。在每个样本中应用质量控制、宏基因组组装以及单覆盖和多覆盖宏基因组分箱过程,总共生成了 63654 个初步 MAG,最小长度为 200 kbp。此外,从初步的MAG中搜索真核生物,发现了17个真核生物基因组,最小基因组大小为4.46 Mbp,其中包括13个真菌基因组、2个阴道毛滴虫 (Trichomonas vaginalis) 基因组和2个小泰勒虫(Theileria parva)基因组。
他们利用来自美国国家生物技术信息中心 (NCBI) RefSeq 基因组数据库的公开原核生物和真菌基因组补充了本项研究的数据,并获得了 1189 个原核生物和 18 个真菌基因组,这些基因组之前是从女性阴道中分离出来的。经过质量过滤后,保留了 972 个高质量的原核生物和 17 个真菌基因组以供进一步分析(图1a)。此外,他们从健康女性的阴道拭子中进行了真菌培养,并获得了 8 种培养真菌,这些真菌纳入本研究的基因组集合。
根据MIMAG(关于宏基因组组装基因组的最少信息)标准,在 19542 个原核生物基因组(即 18570 个 MAG 和 972 个分离基因组)中,分别有 10127、8397 和 1017 个归类为中等质量、高质量和近乎完整的基因组(图1b)。所有原核生物基因组的中位 N50 长度和基因组大小分别为 38.0 kbp和 1.36 Mbp,从近乎完整到中等质量的基因组呈下降趋势(图1c-d)。在 38 个真菌基因组(即 13 个 MAG 和 25 个分离基因组)中,中等完整性和污染分别为 92.7% 和 0.4%,中等基因组大小为 12.7 Mbp(图1e)。
最后,为了进一步扩大病毒群落的范围,他们采用基于特征和同源性相结合的方法对宏基因组组装的重叠群进行病毒鉴定,结果有 14224 个病毒序列达到至少 50% 的完整性(图1a)。基于 CheckV 算法,10.3 % 的病毒序列是完整的病毒基因组,29.3% 的序列为高质量(完整性≥90%),60.4% 的序列为中等质量(图1f)。此外,42.3% 的病毒序列由CheckV 识别为整合的原病毒,并提取了病毒区域。大多数病毒基因组(98.9%)显示出较低程度的基因组污染(<10%)(图1g),宿主与病毒基因比的分析也显示微生物宿主污染较低。病毒序列基因组大小范围为 5.0 kbp 至 417.5 kbp,中位数为 33.5 kbp。高质量病毒的基因组大小大于中等质量病毒;然而,完整病毒的基因组似乎最小(图1h)。
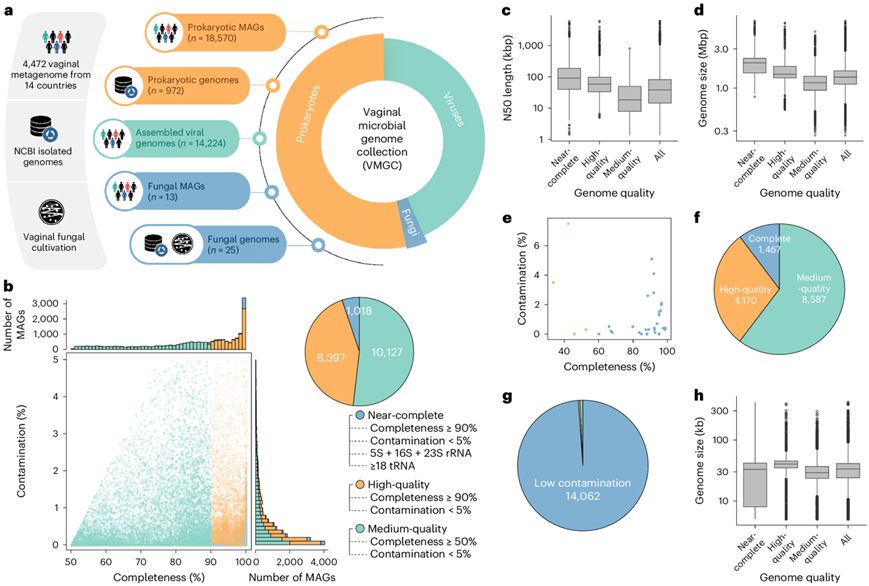
图1. VMGC 的构建和质量评估。
(a) VMGC的构建流程图。(b) CheckM2估计的19542个原核生物基因组的完整性和污染程度。 (c-d) 19542个原核生物基因组的N50长度和基因组大小的分布。(e) BUSCO 估计的 38 种真菌(蓝点)和 4 种寄生虫(橙点)基因组的完整性和污染程度。(f-g) CheckV 估计的 14224 个病毒基因组的完整性和污染程度。(h) 病毒基因组的基因组大小分布。
02
阴道原核生物物种分类概况
他们对 19542 个原核生物基因组进行了全基因组聚类分析,根据 95% 核苷酸相似性阈值产生了 786 个物种级基因组bin (SGB)(以下称为“物种”)。稀疏分析显示物种丰富度未达到饱和,表明存在更多未发现的物种。然而,这些物种很可能主要是稀有物种,因为当仅考虑具有至少两个同种基因组的物种时,物种数量会接近稳定期。786 个物种中约有 41.8%有一个或多个先前从人类阴道中分离出来的基因组,而其余 58.1%的物种只有可用的 MAG(图2a-b)。他们将原核生物物种与 NCBI RefSeq 分离的基因组进行了比较,以将搜索范围扩展到来自其他环境的物种。该分析还鉴定出另外 249 个物种(占所有原核生物物种的 31.7%),它们与之前从其他生境(例如人类胃肠道、口腔或自然环境)培养的基因组具有高度同源性,另有 208 个物种(26.5%)目前尚未培养(图2b)。
阴道原核生物种类分类分析显示,存在 15 门、18 纲、43 目、87 科和 239 属。优势门包括放线菌门(占所有种类的 28.5%)、厚壁菌门_A(21.2%)、拟杆菌门(16.3%),其次是假单胞菌门(6.5%)和芽孢杆菌_C(4.5%)(图2a)。在较低的分类水平上,一些分类单元例如拟杆菌目(占所有物种的 15.0%;主要由普雷沃氏菌属组成)、放线菌目(15.0%;主要为双歧杆菌和放线菌科属)、乳杆菌目(12.6%;主要为乳杆菌和链球菌属)、Tissierellales(9.2%;主要为厌氧球菌和Peptoniphilus属)、红蝽菌目(6.4%;主要为Fannyhessea属)和分枝杆菌目(6.1%;主要为棒状杆菌属)属于主要的进化枝。与人类肠道菌群由拟杆菌属和Lachnospirales(梭菌属、瘤胃球菌属和真杆菌属)组成不同,阴道菌群中仅0.6%的菌属是拟杆菌属,5.3%的菌属是Lachnospirales。
未培养物种广泛分布于不同门类中,尤其是Patescibacteria、Campylobacterota、Spirochaetota和Bacillota_A(图2a)。进一步对含有3个以上未培养物种的9个门类,基于系统发育树计算了系统发育多样性。分析表明,未培养物质平均贡献了这些门系统发育多样性的52.8%(范围从34.3%到83.0%),其中Patescibacteria和Campylobacterota的系统发育多样性比例最高(图2c)。特别是,尽管4个优势阴道门中平均有25.9%的未培养物种,但它们贡献了平均51.8%的系统发育多样性。
一些未培养的物种包含大量基因组,包括 SGB010(称为 BVAB1,一种典型的未培养细菌性阴道病相关细菌,由 461 个基因组组成)、SGB013(391 个基因组)和 SGB015(374 个基因组)。重要的是,这些基因组是从来自不同国家的阴道宏基因组中组装而成的,表明它们在人类群体中广泛存在。此外,他们还分析了宏基因组样本中所有 VMGC 原核生物种的相对丰度。乳杆菌属(如惰性乳杆菌、卷曲乳杆菌和詹式乳杆菌)、双歧杆菌属(阴道双歧杆菌)、普雷沃菌属(双路普雷沃菌和Prevotella timonensis)和Fannyhessea (F. vaginae)是样品的主要成分。一些未分类的细菌,包括BVAB1 (SGB010)、Bifidobacterium sp.(SGB042和SGB058)和Prevotella sp. (SGB013)也属于丰度较高的物种(图2d)。
乳杆菌目(主要是卷曲乳杆菌和惰性乳杆菌)和肠杆菌目(主要是大肠杆菌)对生物膜形成合成的基因丰度有显著贡献,其次是铜绿假单胞菌和BVAB1等潜在病原体(图2e)。他们定量了阴道宏基因组中短链脂肪酸和抗生素抗性基因合成的功能。该分析表明,乳酸杆菌目编码与乙酸和乳酸生物合成相关的基因丰度最高;放线菌目是琥珀酸的潜在主要生产者,而韦荣球菌和Tissierellales分别在生产丙酸和丁酸方面发挥重要作用(图2e)。此外,乳酸杆菌目、放线菌目、假单胞菌目和肠杆菌目表现出最高的抗生素抗性基因丰度。
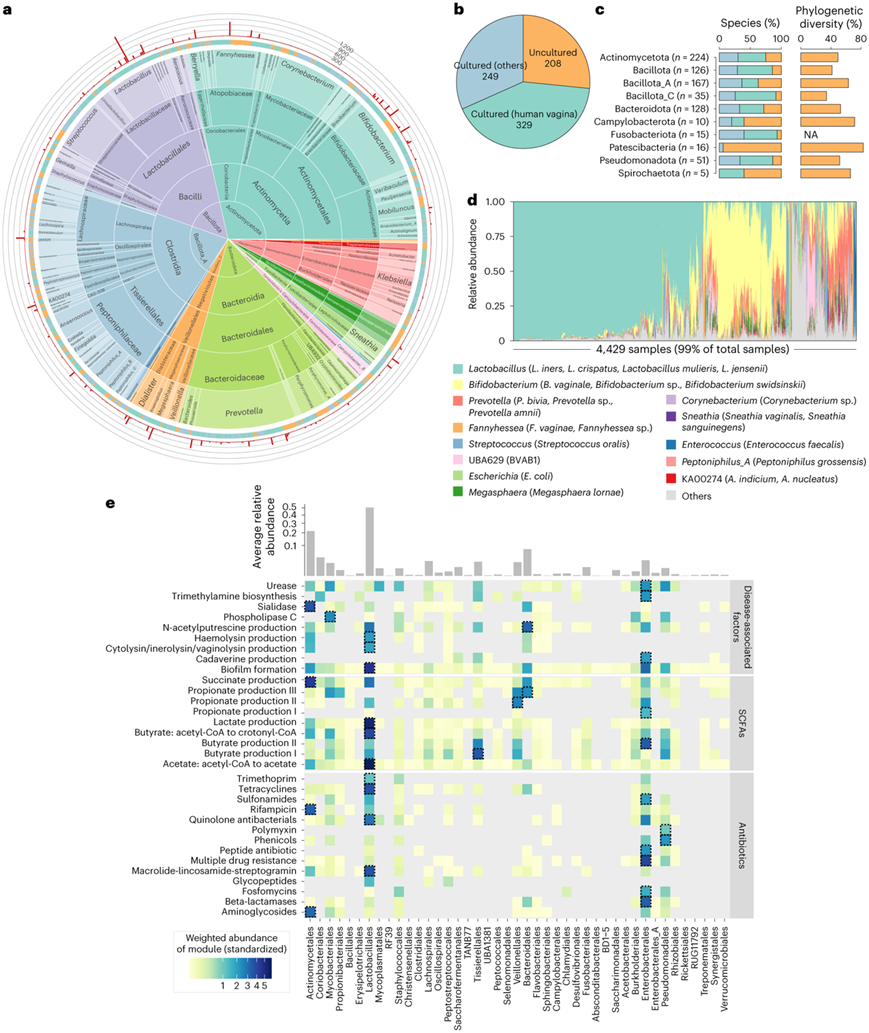
图2. VMGC 中的 786 种原核生物。
(a) 原核生物物种的分类。(b) 未培养物种、从阴道培养的物种和从非阴道部位培养的物种的数量。(c) 不同门中未培养物种、从阴道培养的物种和从非阴道部位培养的物种的比例,以及未培养物种所占的系统发育多样性比例。(d) 4429 个阴道宏基因组中占主导地位的属。(e) 阴道样本中每个目不同功能模块的加权丰度。
03
人类阴道中Saccharofermentanales
Saccharofermentanales是一种流行的阴道细菌,很少在人体其他部位发现,在VMGC中含有几种与细菌性阴道病相关的物种,包括SGB009 (A.indicum,之前称为BVAB2)、SGB034 (M.indolicus,之前称为BVAB3)和SGB080(Amygdalobacter nucleatus,是一种BVAB2样物种)(图3a)。M. indolicus之前已在有限数量的可用基因组中分离和测序,而A. indicium和A. nucleatus 的基因组是在本研究准备期间首次测序的(每个菌在 NCBI 中都有一个完整的基因组)。相比之下,本项研究中的MAG,特别是 SGB009、SGB034 和 SGB080,已产生 12、3 和 1 个近乎完整的基因组,以及大量高质量基因组(分别为 360、98 和 13)。有趣的是,A. indicium和A. nucleatus的估算完整性(完整基因组)保持在 94.1%(图3b)。
他们分析了每个物种的 124 个 通用单拷贝同源物 (USCO),发现它们各自缺少 14 个 USCO 基因(图3c),表明它们的基因组可能在进化过程中丢失了某些关键基因,导致其“完整性”降低。在 SGB034 和其他Saccharofermentanales物种中也观察到了这种现象,所有Saccharofermentanales物种中似乎都有 9 个 USCO 基因缺失,这可能伴随着人类阴道中基因组的明显减少。此外,9 个缺失的 USCO 基因中有 5 个参与了嘌呤从头生物合成(图3c);其他生殖道病原体(如梅毒螺旋体、沙眼衣原体和肺炎支原体)尚未报道缺乏该通路。
他们发现 SGB009 和 SGB080 物种编码 Lsr 型自诱导物-2 (AI-2) 受体,而在其他阴道细菌中几乎不存在这种受体(图3d)。作为跨物种通讯的重要信号分子,AI-2 通过 AI-2 受体在调节多种细菌行为(例如,存活、生物膜形成和毒力相关基因表达)中起着至关重要的作用。在阴道微生物群中,AI-2由LuxS基因催化合成,该基因由双歧杆菌、普氏菌、乳酸杆菌和链球菌等多种细菌编码(图3d-e)。研究表明,具有AI-2递送和内化途径的细菌不仅可以通过AI-2调节其生物膜的形成,而且还能内化外源性AI-2来破坏其他竞争细菌的生物膜形成,这表明SGB009和SGB080物种可以通过内化AI-2来干扰群体感应和其他阴道细菌的生长,从而使它们在竞争中占据优势。
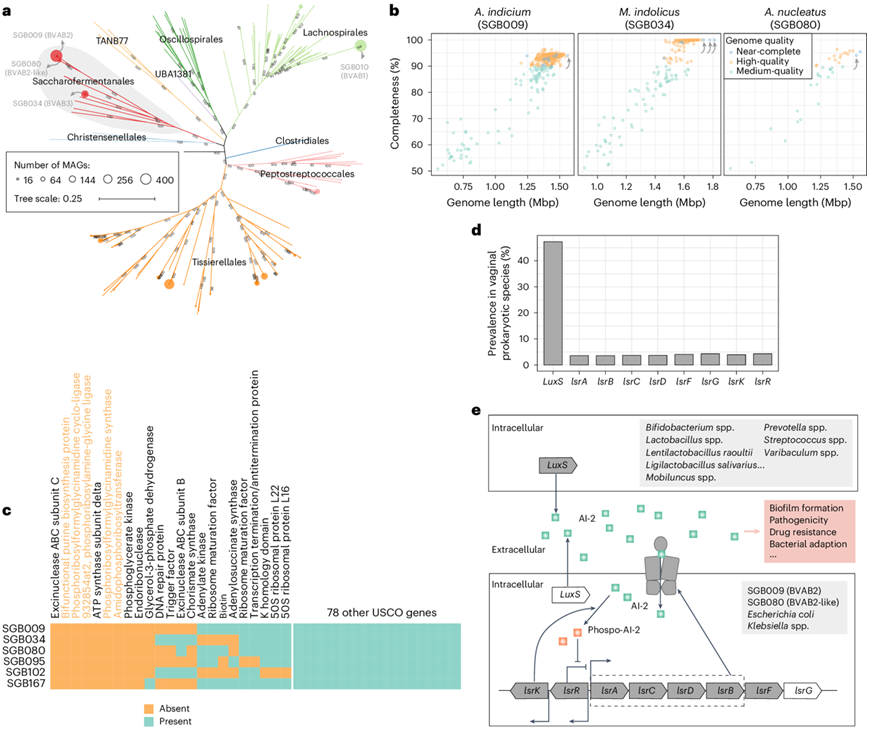
图3. VMGC 中 Saccharofermentanales 成员的基因组特征。
(a) VMGC 中梭菌纲所有物种的系统发育树。 (b) 基于 CheckM2 算法的糖发酵目三个主要成员的基因组完整性得分和基因组大小。(c) 糖发酵目成员中 USCO 基因的存在。(d) VMGC 中所有 SGB 中与 Lsr 型自诱导物-2 (AI-2) 转运系统相关的基因的普遍性。(e) 阴道细菌中存在的 Lsr 型 AI-2 转运系统示意图。
04
阴道病毒的分类和功能多样性
为了探索阴道病毒组的分类内容,他们将VMGC的病毒基因组去重复为4263个物种水平的病毒操作分类单元 (vOTU),核苷酸同一性为95%。稀疏分析表明,尽管非单一vOTU(占所有vOTU的35.5%)已达到饱和,但vOTU的积累曲线在当前病毒基因组数量下并未达到平台期(图4a)。结果显示,VMGC 中的 85.8% vOTU 在其他病毒组数据库中均未发现(图4b)。这些发现表明人类阴道和 VMGC 中的病毒含量存在大量未探索的多样性。
分类学分类显示,66.0%的 vOTU 可以稳健地分配给已知的病毒科,其中 2744 个 vOTU 分配给 13 个原核病毒科,70 个 vOTU 分配给 7 个真核病毒科(图4c)。VMGC 中观察到最多的科是长尾噬菌体科和肌尾噬菌体科,其次是乳头瘤病毒科、Rountreeviridae、短尾噬菌体科、Quimbyviridae、Autographiviridae、p-crAss-like和其他科。唾液病毒科和肌尾噬菌体科是肠道和口腔等人体部位最主要的病毒科,然而,另一种在人类肠道中占主导地位的病毒家族-微病毒科,很少在阴道中发现。此外,VMGC中86.4%的vOTU至少有一个预测的原核宿主(图4c)。在门水平上,阴道病毒的预测宿主以放线菌门、拟杆菌门、杆菌门和Bacillota_A为主。
他们根据 4263 个 vOTU 在蛋白质水平上的基因组相似性构建了系统发育树(图4d)。该树显示病毒倾向于按科水平分类和潜在宿主关系聚类。这一发现与从口腔病毒目录获得的结果基本一致,并表明宿主适应是驱动人相关病毒基因组进化的重要因素。预测长尾噬菌体科 vOTU 会感染放线菌门、芽孢杆菌门、Bacillota_A 和其他细菌(图4c)。长尾噬菌体科 包含许多病毒(占所有 Siphoviridae 病毒的 20.6%),预测这些病毒会感染多个原核生物门的宿主;但这种现象在其他科的病毒中很少观察到(12个原核病毒科平均为2.4%),表明阴道长尾病毒科成员的宿主范围相对较广。预测肌尾噬菌体科会感染Bacillota_A、Bacillota、Pseudomonadota等,但Rountreeviridae和Autographiviridae似乎分别特定于Bacillota和Bacillota_C的感染。值得注意的是,拟杆菌噬菌体在系统发育树中明显集中(图4d)。具体来说,在感染拟杆菌的520种噬菌体中,大多数(86.2%)是未分类的病毒,表明人类阴道中存在许多以前未知的拟杆菌噬菌体类群。
为了详细说明阴道病毒组的功能概况,他们利用KEGG数据库注释了所有vOTU的功能,结果13.9%的病毒基因具有KEGG直系同源注释。在这些注释的基因中,49.0%参与遗传信息处理,19.9%是病毒辅助代谢基因(图4e)。重点关注VMGC中最普遍的辅助代谢基因,其中许多与肽聚糖、核苷酸、氨基酸、叶酸和硫的代谢有关(图4f)。
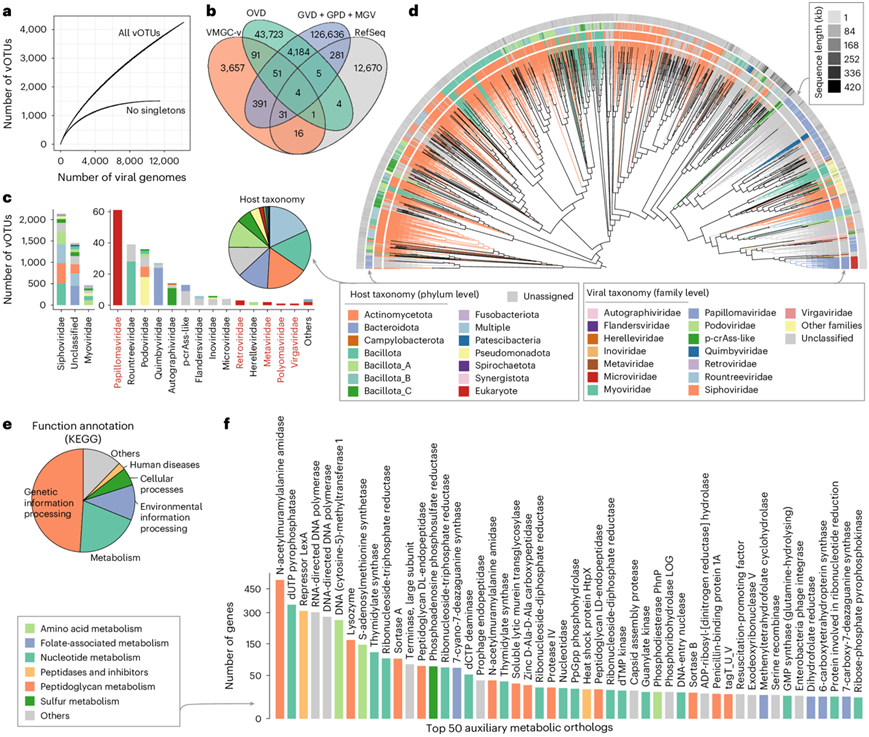
图4. VMGC 中病毒种群的特征。
(a) 随着病毒基因组数量的增加,vOTU 的积累曲线。(b) 几个大型病毒基因组目录中病毒种类的重叠。(c) 4263 个 vOTU 的宿主门分布。(d) 蛋白质组树显示 4263 个 vOTU 之间的关系。(e) 4263 个 vOTU 中 KEGG 注释基因的功能分布。 (f) 4263个 vOTU 中前 50 个辅助代谢直系同源物的基因数量。
05
阴道真核病毒的特征
在鉴定的 70 个真核 vOTU 中,他们特别关注乳头瘤病毒科病毒,因为人乳头瘤病毒 (HPV) 在阴道中分布广泛且与宫颈癌等疾病有关。VMGC 中的几乎所有乳头瘤病毒都具有完整或高质量的基因组,大多含有完整的L1基因(编码主要衣壳蛋白)以进行 HPV 分型(图5a)。他们根据L1基因将 61 个乳头瘤病毒科 vOTU 分为 58 种 HPV 类型(包括 5 种未鉴定的类型),其中 52 种类型之前是在女性阴道中发现的,6 种类型之前是在人类阴茎和皮肤拭子中发现的。同样,对 61 个乳头瘤病毒科 vOTU和 NCBI RefSeq 乳头瘤病毒进行系统发育分析表明,vOTU 代表了人阴道乳头瘤病毒的主要系统发育进化枝(图5b)。此外,基于基因组,发现对于某些 HPV 类型,它们的基因组进化关系具有明显的区域分层(以 HPV 类型 52 和 58 为例)(图5c)。基于 VMGC 的阴道宏基因组组成分析展示了其在识别宫颈病变或癌症患者中某些 HPV 类型的富集方面的潜力。总的来说,这些发现凸显了 VMGC 乳头瘤病毒科病毒作为后续 HPV 研究的可代表参考目录的价值。
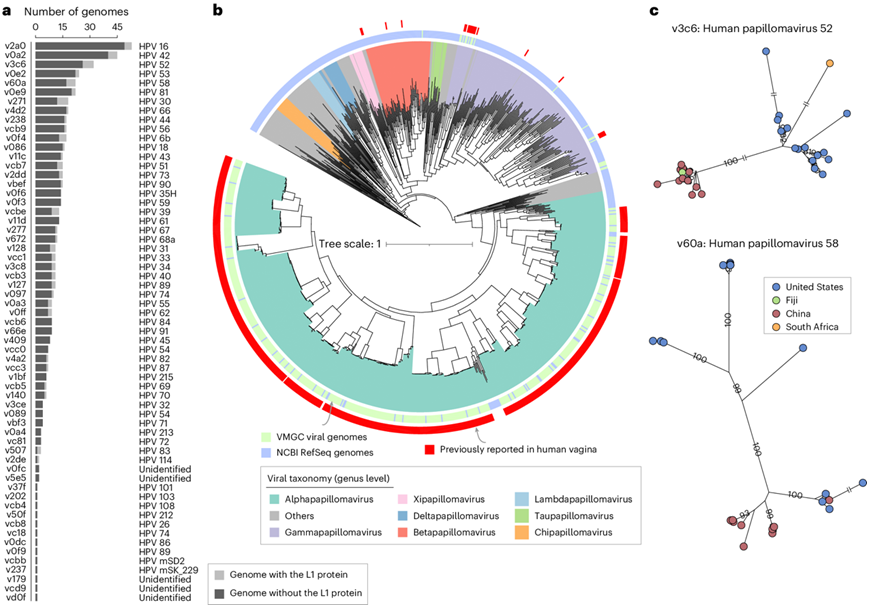
图5. VMGC 中乳头瘤病毒科成员的特征。
(a) 标注有乳头瘤病毒科 (Papillomaviridae) 的 61 个 vOTU 的基因组数量和 HPV 分型。 (b) 基于 VMGC 和 NCBI RefSeq 数据库中所有乳头瘤病毒科基因组的 L1 蛋白的系统发育树。(c) 基于 VMGC 中 HPV52 和 HPV58 基因组的系统发育树。
06
阴道微生物基因目录及与 VIRGO 的比较
为了探索 VMGC 的基因含量,他们从所有原核生物、真菌和病毒基因组中按 50%(VMGC-50)、90%(VMGC-90)和 95%(VMGC-95)氨基酸同一性对大约 2600 万个蛋白质编码基因进行了聚类,生成了三个目录,分别包含 595219、1415799 和 1786695 百万个非冗余基因。稀疏分析显示,VMGC-95 和 VMGC-90 的积累曲线尚未达到平台期,而 VMGC-50 的曲线已接近饱和(图6a),这表明 VMGC 代表了人类阴道微生物组的基因空间不足但基因家族空间相对饱和。此外,他们将 VMGC-90 的基因含量与阴道非冗余基因目录 (VIRGO)进行了比较,后者代表了 62 万个基因,氨基酸同一性为 90%(称为 VIRGO-90)(图6b)。此外,将阴道宏基因组与阴道微生物基因和基因组目录进行比对,发现来自 14 个不同国家的样本的平均比对率分别为 74.5% 和 83.8%,略高于 VIRGO(平均 71.7%)(图6c)。欧洲或北美样本的比对率高于中国、斐济或南非样本,表明后者地区 VMGC 基因的代表性相对较低。
他们进一步在 VMGC-90 水平上比较了原核生物、真菌和病毒的基因含量。超过一半(53.7%)的病毒基因与原核生物共存(图6d),可能是因为这两个界中遗传物质交换频繁,而真菌与原核生物/病毒之间只有少数基因共存。他们还比较了原核生物特异、真菌特异和病毒特异基因的功能。如预期的那样,病毒特异基因主要编码典型的病毒酶,涉及遗传信息处理(例如 DNA 复制和修复以及转录)和原核生物防御系统,而原核生物和真菌特异基因具有更多的代谢相关基因。

图6. VMGC中微生物基因的特征。
(a) 非冗余蛋白质随取样蛋白质数量增加的积累曲线。(b) VMGC-90与VIRGO-90之间蛋白质的重叠。(c) 对应于VMGC-90的基因目录。(d) VMGC-90中原核生物、真菌和病毒蛋白质的重叠。
+ + + + + + + + + + +
结 论
本项研究利用宏基因组测序数据和真菌培养建立了VMGC,包含 33804 个微生物基因组,涵盖 786 个原核生物物种、11 个真菌物种和 4263 个病毒操作分类单元。该集合大大丰富了基因组多样性,尤其是对于流行的阴道病原体,例如 BVAB1(一种未培养的细菌性阴道病相关细菌)和杏Amygdalobacter spp.(BVAB2 和相关物种)。利用 VMGC,描述了原核生物的功能特征,特别是Saccharofermentanales(一种尚未充分研究但普遍存在的目)以及原核和真核病毒,从而深入了解了它们的生态位适应性和在阴道中的潜在作用。VMGC 是研究阴道微生物群及其对阴道健康影响的宝贵资源。
+ + + + +



