

 English
English文献解读|Nat Aging(17.0):老年人虚弱的肠道微生物特征和循环代谢组学特征
✦ +
+
论文ID
原名:Gut microbial features and circulating metabolomic signatures of frailty in older adults
译名:老年人虚弱的肠道微生物特征和循环代谢组学特征
期刊:Nature Aging
影响因子:17.0
发表时间:2024.07.25
DOI号:10.1038/s43587-024-00678-0
背 景
虚弱是亚最佳衰老的多维指标,反映了多个生理系统的累积衰退。尽管据报道肠道微生物群发生了与年龄相关的变化,但它们在健康衰老中的作用仍不清楚。
实验设计
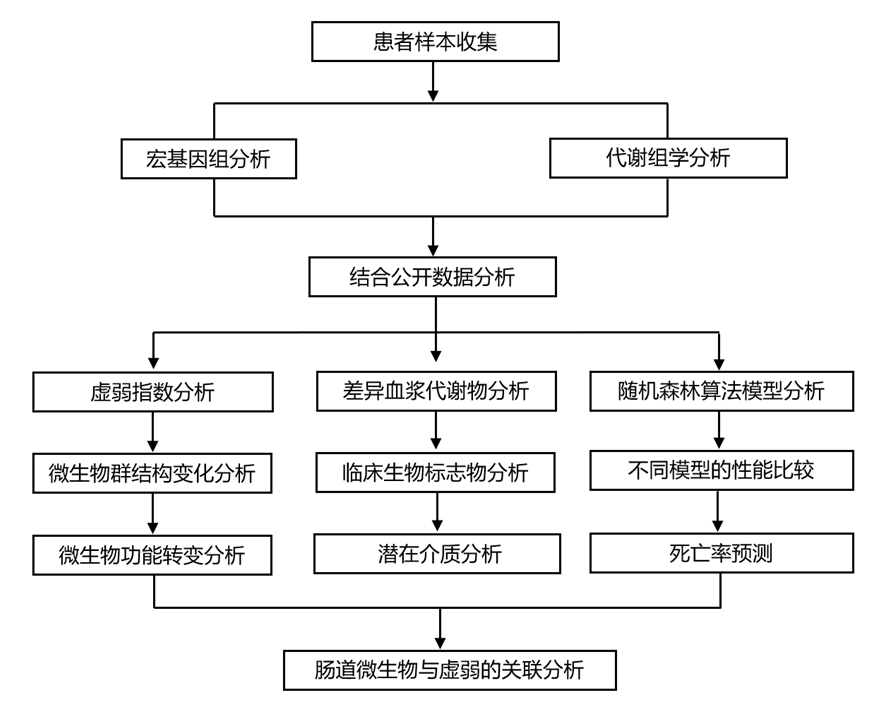
结 果
01

虚弱者肠道微生物群的结构变化
研究团队开展了一项如皋纵向老龄化研究 (RLAS),共纳入了 1821 名 RLAS 参与者(55% 为女性),年龄范围为 62-96 岁,并用虚弱指数 (FI)进行评估(图1)。在这些参与者中,253 人(13.9%)归类为不虚弱(FI ≤ 0.1),962 人(52.8%)归类为轻度虚弱(0.1 < FI ≤ 0.2),413 人(22.7%)归类为中度虚弱(0.2 < FI ≤ 0.3),193 人(10.8%)归类为重度虚弱(FI > 0.3)。排除有胃肠道手术史、取样前服用抗生素或未提供粪便样本或宏基因组测序深度不足的参与者后,总共有 1448 名参与者纳入肠道宏基因组分析。该队列的肠道宏基因组谱主要由拟杆菌(52.4%)和厚壁菌门(35.3%)组成。四个虚弱组之间的 alpha 多样性指数没有显著差异。然而,不同虚弱严重程度的参与者的整体肠道微生物结构存在显著差异(图2a)。进一步的研究表明,与人口统计学因素(年龄和性别)、临床实验室结果和自我报告的生活方式或健康指标相比,虚弱严重程度在微生物分类学变异中所占比例更大(图2b)。亚组分析显示,各衰弱组和非衰弱组之间的β多样性差异仅在女性参与者中显著。相反,在男性参与者中没有观察到这种差异。
MaAsLin 分析显示,在调整混杂因素(即年龄、性别、体重指数BMI、吸烟、饮酒、教育和体力活动水平)后,18 种微生物与虚弱严重程度显著相关(图2C)。在虚弱进展过程中,13 种细菌(Bacteroides eggerthii、Clostridium bolteae、Clostridium clostridioforme、Clostridium innocuum、Erysipelatoclostridium ramosum、Flavonifractor plautii、Hungatella hathewayi、Megamonas funiformis、Megamonas hypermegale、Parabacteroides merdae、Prevotella sp. 885、Proteobacteria bacterium CAG:139 和Ruminococcus gnavus)的相对丰度显著增加,而 5 种细菌(Adlercreutzia equolifaciens、Faecalibacterium prausnitzii、Oscillibacter sp. 57 20、Roseburia sp. CAG:471 和Ruminococcus callidus)的相对丰度降低。三种细菌(即E. ramosum、F. prausnitzii和R. gnavu s)之间的关联与虚弱严重程度受性别影响显著(图2d)。
随后,他们结合来自中国和美国的数据集研究微生物种类与虚弱的关联(图2e)。在中国的数据集中,鉴定出上述 17 种与虚弱症相关的细菌,其中 7 种在调整年龄、性别、BMI、吸烟和饮酒状况以及运动水平后,表现出与虚弱症严重程度一致且显著的关联。在美国的数据集中,检测到了 13 种与虚弱症相关的细菌,其中 5 种与虚弱症严重程度保持显著关联,两种表现出较低程度的显著关联,值得注意的是, C. innocuum、C. bolteae、C. clostridioforme和R. gnavus的相对丰度在所有虚弱组中持续增加。

图1. 研究人群、检测和分析策略。
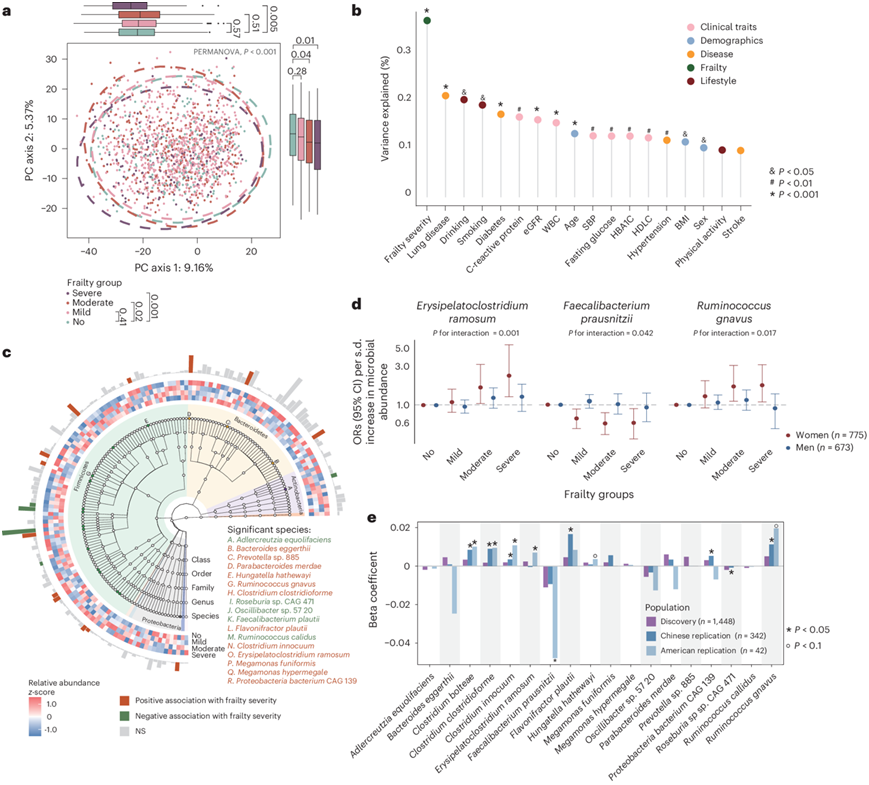
图2. 虚弱严重程度与整体肠道微生物组成和单个肠道微生物种类的关系。
(a) PCA 显示基于物种水平 Aitchison 差异性的不同虚弱严重程度群体的肠道微生物组成。(b)定量虚弱严重程度、人口统计和生活方式因素以及健康相关特征解释的分类学变异比例。(c) 分类特征以系统发育树的形式呈现,突出显示与虚弱严重程度显著相关的物种。(d) 与虚弱严重程度相关的性别特异性微生物特征。(e) MaAsLin 分析用于评估虚弱严重程度与发现队列和外部数据集中的肠道微生物特征之间的关联。
02
与虚弱相关的血浆代谢组学特征
他们利用基于核磁共振 (NMR) 的平台对 1771 名参与者血浆样本进行了代谢组学分析。代谢组学数据的偏最小二乘判别分析 (PLS-DA) 导致无衰弱、轻度衰弱、中度衰弱和重度衰弱组之间的代谢特征明显分离(图3a),并且 81 种代谢物的投影变量重要性 (VIP) 评分高于 1.0 有助于观察到的分离并包括在以下分析中(图3b)。多元变量线性回归分析显示,17 种代谢物与衰弱严重程度显著相关(图3c)。检测的最小高密度脂蛋白(即 HDL4)中的成分始终与虚弱严重程度呈负相关,其中 HDL4 中的载脂蛋白 A1(H4A1)表现出最大的关联。随着虚弱严重程度的进展,亮氨酸、赖氨酸和乙酰乙酸的循环水平下降,而葡萄糖和甘油的水平升高(图3b)。此外,与不虚弱者相比,血浆中肠道微生物来源的三甲胺氧化物 (TMAO) 水平每增加一个标准差(s.d.),中度虚弱的几率就会增加 30%,重度虚弱的几率就会增加 39%(图3c)。
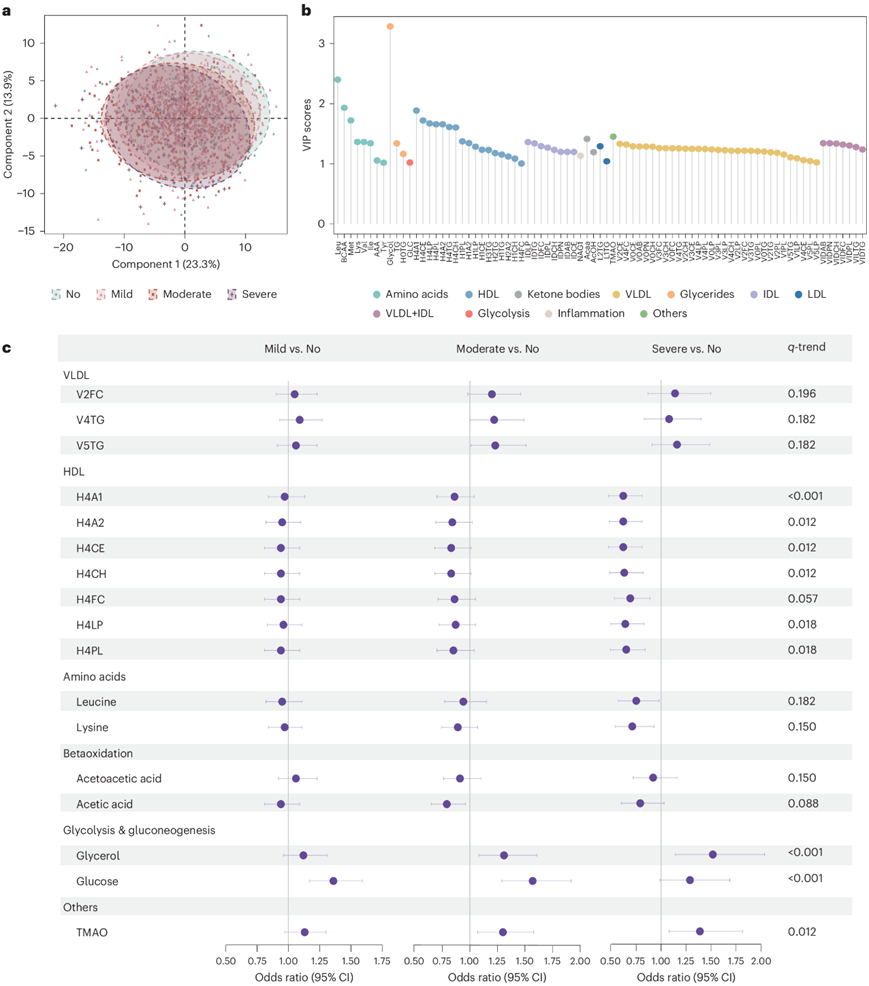
图3. 不同虚弱严重程度的参与者的血浆代谢物有所不同。
(a) 不同虚弱程度参与者的血浆代谢组谱的 PLS-DA。(b) VIP 评分高于 1.0 的代谢物。(c) 17 种选定代谢物与虚弱严重程度显著关联。
03
通过临床生物标志物和代谢物的调节作用
接下来,他们使用上述与虚弱症相关的临床生物标志物和代谢物作为潜在介质进行了中介分析。共确定了 34 个标志来介导微生物与虚弱症严重程度之间的关联,主要通过全身炎症、异常的葡萄糖和脂质代谢以及肾功能障碍(图4a)。例如,F. plautii通过能量代谢指标与虚弱严重程度呈正相关,例如空腹血糖、甘油和静息脉率(图4b)。同样,P. bacterium CAG:139 与虚弱严重程度的关联显著由全身炎症调控,其中白细胞和单核细胞计数分别介导 14.4% 和 11.0% 的关联。此外,C. bolteae与虚弱严重程度之间的关联由肾功能障碍调控,其中 14.4% 通过 β2-微球蛋白调控(图4c)。一些微生物与虚弱的严重程度通过多个系统功能障碍存在关联。比如,H. hathewayi与虚弱程度呈正相关,这种关联通过以下途径介导:系统性炎症(由白细胞数量反映,介导效应为6.7%,P值为0.008),肾功能障碍(通过β2-微球蛋白反映,介导效应为12.0%,P值小于0.001),以及能量代谢紊乱(通过甘油反映,介导效应为8.4%,P值为0.002)。总体而言,这些结果表明,系统性炎症、肾功能障碍以及葡萄糖和脂质代谢在肠道微生物与虚弱之间的关联中起着重要的中介作用。

图4. 临床生物标志物和循环代谢物在肠道微生物种类和虚弱严重程度之间的调节。
(a) 平行坐标图显示在q值 < 0.2 时显著的 34 种临床生物标志物和循环代谢物的药物关联。(b) F. plautii的相对丰度与虚弱严重程度的关联由空腹血糖、甘油和静息脉搏率介导。(c) P. bacterium CAG:139 的相对丰度与虚弱严重程度的关联由白细胞和单核细胞计数介导, C. bolteae的相对丰度与虚弱严重程度的关联由 β2-微球蛋白介导。
04
利用多组学特征对虚弱进行重新分类
为了评估与衰弱相关的微生物和代谢组学特征增强的预测能力,他们将参与者随机分成训练集(80%)和验证集(20%)。他们实施了一个强大的验证过程,使用 100 次五重交叉验证迭代来确保该模型的可靠性和稳定性。通过将与衰弱相关的微生物群和代谢物添加到之前仅包含传统风险因素(如人口统计和生活方式)的重新分类模型中,显著增强了该模型区分严重衰弱参与者与没有或较轻衰弱参与者的能力[受试者工作特征曲线下面积(AUROC)从 0.57提高到 0.74(图5a)。然而,在区分非衰弱参与者和衰弱参与者方面没有观察到增强(图5b)。值得注意的是,在区分严重虚弱的参与者和非虚弱的参与者时,所有包含传统风险因素的模型(无论是否包含肠道微生物和/或血浆代谢组学特征)都表现出了较好的鉴别能力(所有 AUROC ≥ 0.82)(图5c)。
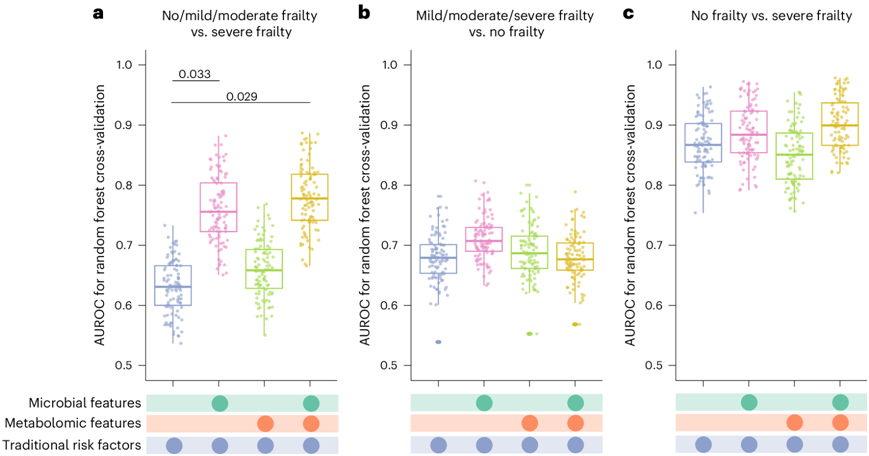
图5. 使用随机森林算法建立模型。
(a) 模型用于识别严重衰弱症参与者与无衰弱症、轻度衰弱症或中度衰弱症参与者的AUROC 值。(b) 模型用于识别无衰弱症参与者与轻度、中度或严重衰弱症参与者的AUROC 值。(c) 模型用于识别严重衰弱症参与者与无衰弱症参与者的AUROC 值。
05
基于组学的虚弱指数的死亡率预测
为了全面评估这些与虚弱相关特征的总体关联,他们使用缺陷累积方法构建了复合微生物 FI(FI-微生物群)和代谢组 FI(FI-代谢物)模型。正如预期的那样,表现出 FI-微生物群或 FI-代谢物评分升高的参与者更有可能具有更高的传统 FI(FI-传统)评分。此外,他们建立了一个组合 FI(FI-combined)模型,以全面涵盖上述三个 FI 中的所有组成部分。生存分析显示,所有 FI 都与死亡率显著相关(图6a-d),进一步调整年龄和性别并没有显著改变结果(模型 1)(图6e)。值得注意的是,FI 微生物群与 2 年死亡率的关联性比 FI 代谢物、FI-传统甚至 FI 组合更强。此外,在同时调整年龄、性别、FI-传统和 FI 代谢物的模型中,FI 微生物群仍然与死亡率显著相关(模型 2)(图6e)。
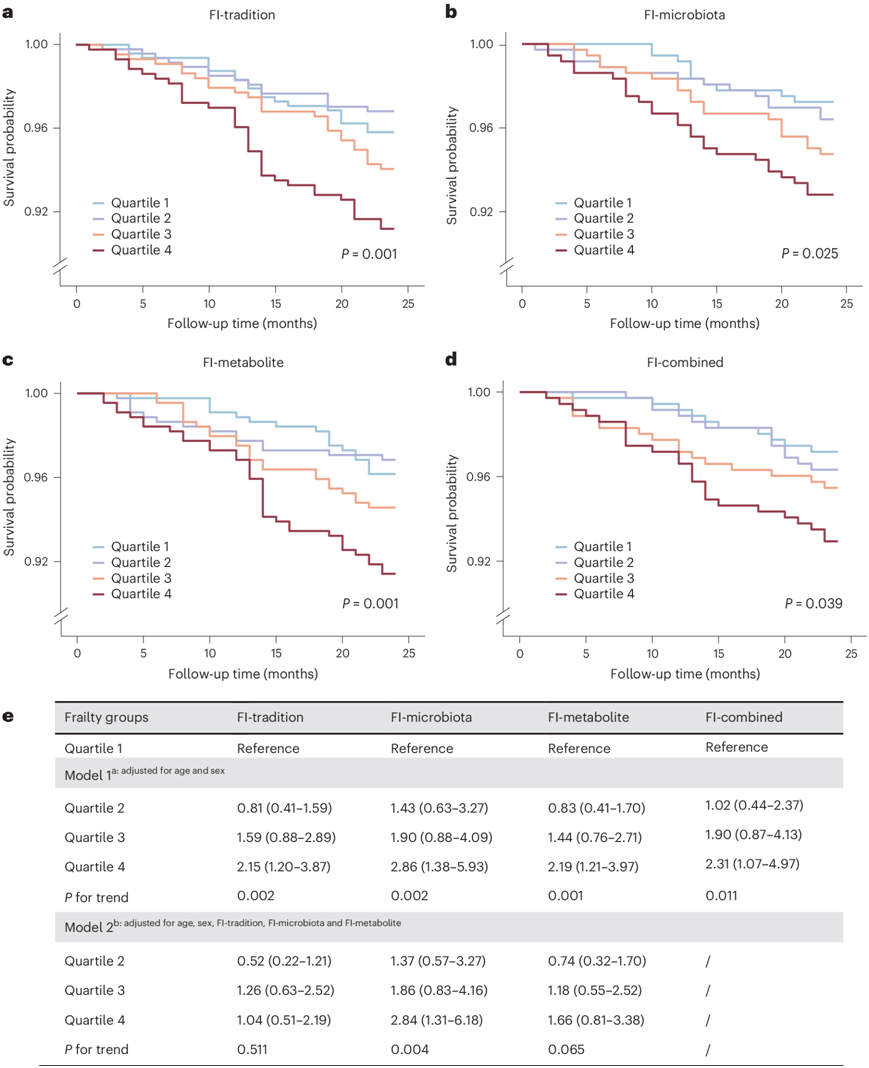
图6. 传统、基于组学和组合的 FI 作为 2 年全因死亡率的预测指标。
(a-d) Kaplan–Meier 曲线显示不同 FI 与死亡时间的关系。(e) 不同 FI 与死亡率的 Cox 回归分析。
+ + + + + + + + + + +
结 论
本研究使用肠道宏基因组测序数据和血浆代谢组学数据进行多组学分析,发现 18 种微生物和 17 种代谢物随虚弱严重程度而变化,且在女性中观察到更强的联系。血浆甘油水平、白细胞计数和肾功能部分介导了这些关联。从 FI 得出的综合微生物评分可显著预测 2 年死亡率,凸显了基于微生物群的策略对老年人风险分层的潜力。
+ + + + +



