

 English
English文献解读|Sci Adv(13.6):阿尔茨海默病中的人脑糖型共调节网络和聚糖修饰改变
✦ +
+
论文ID
原名:Human brain glycoform coregulation network and glycan modification alterations in Alzheimer’s disease
译名:阿尔茨海默病中的人脑糖型共调节网络和聚糖修饰改变
期刊:Science Advances
影响因子:13.6
发表时间:2024.04.05
DOI号:10.1126/sciadv.adk6911
背 景
阿尔茨海默病 (AD) 是最常见的痴呆症,其病理特征是大脑中淀粉样蛋白 - β (Aβ) 斑块和 tau 神经原纤维缠结的积累。全基因组关联研究 (GWAS) 已确定了 40 多个 AD 遗传风险位点,转录组和蛋白质组分析揭示了 AD 大脑中 RNA 和蛋白质表达变化比 Aβ 和 tau 积累更广泛。然而,仅 DNA 变异和 RNA 或蛋白质水平变化不足以解释 AD 病理学的所有方面,但目前对人脑中糖基化蛋白质或糖型及其在AD中的改变的了解仍然有限。
实验设计
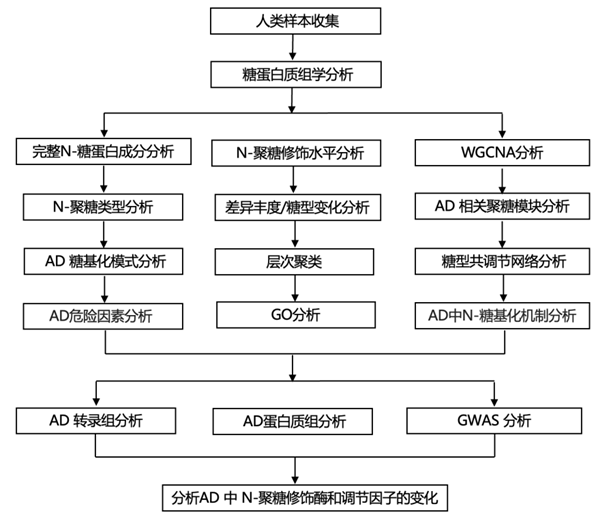
结 果
01

完整的糖蛋白组学揭示了 AD 和对照大脑的糖蛋白组学景观
为了表征人脑糖蛋白组及其在 AD 中的变化,研究者团队建立并优化了基于质谱的完整糖蛋白组学平台,用于大规模同时分析脑样本中的糖蛋白、糖型、糖位点和位点特异性聚糖组成。他们鉴定了 12176 个独特的 N-糖肽,在 NXS|T (X ≠ P) 序列子处附着有 N-聚糖,对应于 10731 个独特的 N-糖型,在1184种独特N-糖蛋白中的2544个独特N-糖基上具有164种不同N-聚糖组成(图1A-D)。该数据集代表了迄今为止人类 AD 和对照大脑最大的糖蛋白质组数据集,具有位点特异性 N-聚糖和糖型信息。将糖蛋白组数据集与来自同一队列的蛋白质组谱数据进行比较,发现1184个糖蛋白中有348个(约29%)是完整的糖蛋白组学鉴定出的(图1E),这表明本项研究的糖蛋白组学工作流程能够分析出一些在整体蛋白质组谱中可能无法检测到的蛋白质子集。正如预期的那样,糖蛋白组数据集中富集了质膜和细胞外蛋白、溶酶体和内质网管腔蛋白、神经元和突触膜蛋白以及受体、离子通道和细胞黏附分子。本研究中发现的约51%的N-糖蛋白和41%的N-糖苷位点与之前使用基于去糖基化的糖蛋白组学方法中检测到的数据相重叠(图1E-F)。
将本项研究完整的糖蛋白组学数据集与UniProt数据库进行匹配显示,识别的2544个N-糖基中有65%在数据库中进行了注释或预测(图1G)。目前的研究为超过950个UniProt预测的N-糖位点提供了实验证据,并确定了超过880个以前未在UniProt中报道的N-糖位点。本项研究的糖蛋白组学分析在对照组中鉴定出7772种N-糖型,在AD患者中鉴定出8196种N-糖型,其中5237种N-糖型在对照组和AD患者大脑中重叠(图1D)。
他们在对照组和AD大脑中鉴定了164种N-聚糖组成,并将其分为7种聚糖类型:寡甘露糖、寡甘露糖、壳二糖核心、未修饰的复合/杂化(既非岩藻糖化也非唾液酸化)、仅岩藻糖化、仅唾液酸化和唾液酸-岩藻糖化(唾液酸化和岩藻糖化)聚糖。在对照大脑中,约36%的N-糖型携带寡甘露糖型聚糖,约41%携带含岩藻糖化的聚糖(图1H),这与之前的糖蛋白组学和糖组学研究的结果一致。AD大脑中仅唾液酸化的糖型、唾液酸岩藻糖基化的糖型和总唾液酸化的糖型的百分比显著降低,而仅岩藻糖基化的糖型和未修饰的复合/混合糖型的百分比在AD中显著增加(图1H)。为了深入了解N-聚糖分支,他们根据聚糖组合物中触角HexNAc残基的数量(即HexNAc残基总数-2)将N-聚糖分为不同的分支类别(图1H)。注意到这些分支类别是假定的结构分配,因为单个聚糖组合物可能代表多个聚糖结构和分支类型。AD 大脑中携带总共 6 个 HexNAc 残基(指定为四触角聚糖)或 > 6 个 HexNAc 残基(高度支化或伸长的聚糖)的聚糖的百分比显著降低(图 1H)。
他们在对照组大脑中鉴定了1952个N-糖基,在AD大脑中鉴定了2114个N-糖基,其中1522个N-糖基是对照组和AD大脑共有的(图1C)。在对照组大脑中,观察到大约57%的N-糖基是寡甘露糖聚糖,约60%是含岩藻糖化的聚糖,43%是含唾液酸的聚糖,约13%是未修饰的复合物/杂化聚糖,约9%是寡甘露糖聚糖,约5%是壳二糖核心型聚糖(图1I)。每个N-糖苷元并没有单独归类为一种糖基类型,因此,如果一个N-糖苷元与几个不同的糖基连接,那么它可以算作多种糖基类型。在AD患者中,唾液酸-岩藻糖化糖苷和总唾液酸化糖苷的百分比显著降低,而仅岩藻糖化糖苷的百分比显著增加(图1I)。在对照组和AD的大脑中,超过一半的N-糖蛋白是在一个糖基上由N-糖基化的,而大约10%是在体内由5个或更多的N-糖基化位点由多重糖基化的(图1J)。在对照组和AD大脑中,每个糖蛋白的体内N-糖苷元平均数量均为2.1。糖蛋白组学分析显示,修饰每个糖苷的不同N-聚糖的数量存在广泛的异质性(图1J)。在对照组和AD脑中,大约45%的总糖位点含有一种聚糖,而约18%的糖位点含有5种以上的聚糖,约1%的糖位点含有30种以上的聚糖(图1J)。
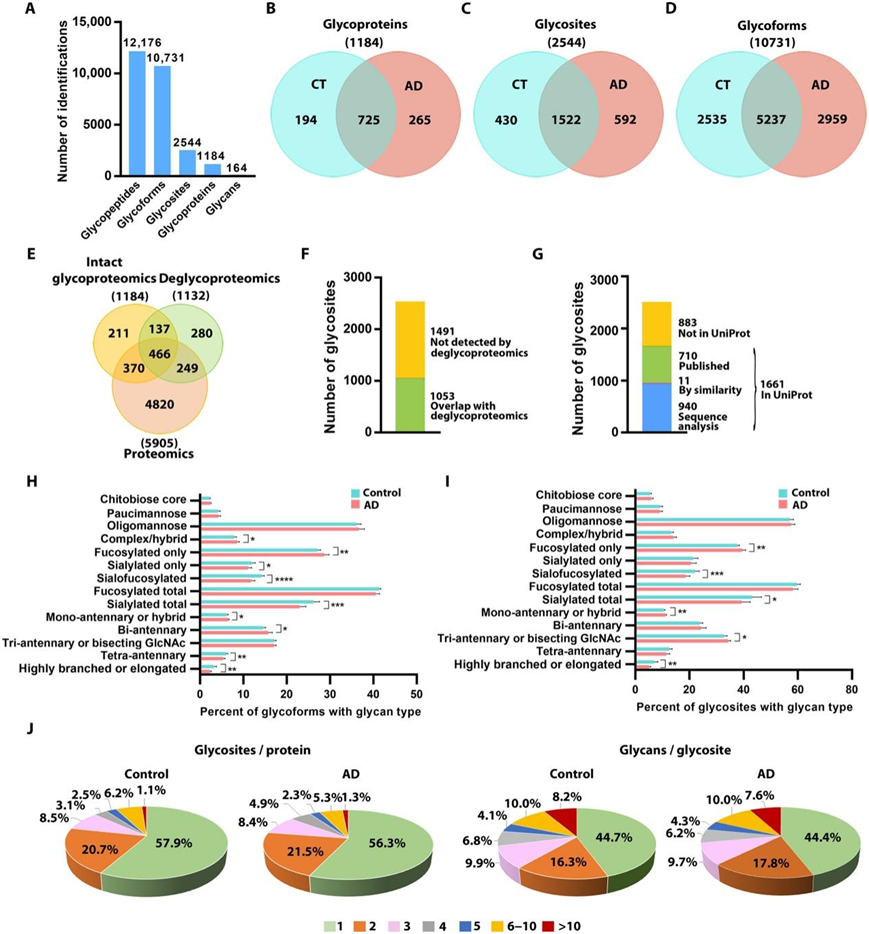
图1. 对人类 AD 和对照大脑进行基于完整糖肽的糖蛋白组学分析。
(A) 本研究中确定的独特 N-糖肽、N-糖型、N-糖蛋白、N-糖位点和 N-聚糖成分的总数。(B-D) AD 与对照 (CT) 大脑中已识别的 N-糖蛋白、N-糖位点和 N-糖型的比较。(E) 完整糖蛋白组学鉴定的 N-糖蛋白与蛋白质组学和去糖蛋白组学检测的蛋白质的比较。(F) 已识别的 N-糖位点与去糖蛋白组学检测到的重叠。(G) 已识别的 N-糖基化位点与带注释的 N-糖基化位点的 UniProt 数据库的匹配。 (H-I) AD 和对照大脑中含有指定聚糖类型的已识别 N-糖型和 N-糖位点的分布。(J) 饼图显示 AD 和对照大脑中每个蛋白质具有指定数量的体内 N-糖位点(宏观异质性)的糖蛋白的比例以及每个位点具有指定数量的 N-糖基团(微观异质性)的糖蛋白的比例。
02
AD 相关蛋白的位点特异性糖基化谱和危险因素
本项研究的糖蛋白组学分析在AD和对照组脑组织中映射了一系列基因组关联研究(GWAS)鉴定的AD风险因子(例如ABCA7、ACE、ADAM10、CLU、CNTNAP2、EPHA1、PLD3、SORL1和TM2D3)以及其他与AD相关的蛋白质(例如,tau、nicastrin和LRP1)的位点特异性N-糖基和N-糖基型(图 2A-B)。检测到的AD相关蛋白的糖基化谱显示了广泛的糖基和糖型多样性,一些蛋白(如ABCA7和EPHA1)只有一个糖型,在一个糖基上携带一个糖基,而其他蛋白(如CLU和LRP1)有80多个糖型,在多个糖基上携带30个或更多的糖基(图 2A-B)。CLU(丛生蛋白,也称为载脂蛋白J)是AD的第三大重要危险因素,具有细胞外伴侣功能。他们在对照组的大脑中共鉴定出88种CLU糖型,分别携带4个糖基的60种不同聚糖;在AD患者的大脑中共鉴定出104种CLU糖型,分别携带4个糖基的67种聚糖,其中大多数糖型携带岩藻糖化和/或唾液酸化聚糖。这些结果揭示了26种AD特异性CLU糖型和10种对照特异性CLU糖型(图2A-B),表明AD与CLU N-糖基化异常有关。

图2. AD 相关蛋白和危险因素的位点特异性聚糖和糖型图谱。
(A) 条形图显示了与 AD 和对照 (CT) 大脑中每个 AD 相关蛋白和危险因素相对应的已识别 N-糖位点、N-聚糖组成和 N-糖型的数量。(B) 与对照大脑相比,AD 中 CLU、tau、TM2D3 和 SORL1 的位点特异性糖基化模式。
03
AD 中唾液酸化和 N-聚糖分支或伸长减少以及甘露糖基化和 N-聚糖截短升高
为了进一步表征 AD 中蛋白质 N-聚糖修饰及其变化,他们对不同聚糖类型的糖肽强度进行了定量分析,以确定 AD 和对照病例的个体脑样本中每个聚糖类别的聚糖修饰的相对丰度(图3A-C)。低聚甘露糖聚糖对蛋白质的修饰在人脑中非常丰富,相对丰度约为 49%(图 3A)。在大脑蛋白中含量最高的7个N-聚糖中,有5个由H5N2到H9N2组成,对应的是含有2个核心GlcNAc和5 ~ 9个甘露糖残基的寡糖结构(Man5到Man9聚糖),其中H5N2是健康人脑中含量最高的N-聚糖,相对丰度约为21%(图 3C)。在前7位含量最高的糖基中,另外两个具有H3N5F1和H3N4F1的组成,这与之前从脑糖蛋白释放的N-糖基的糖组学分析的结果一致。H5N2、H3N5F1和H3N4F1修饰丰度在AD中显著升高,而H9N2修饰丰度在AD中显著降低(图3C)。
定量分析表明,在对照组的大脑中,岩藻糖化和唾液化的相对丰度分别为39%和19%(图3A)。AD脑中的唾液酸化丰度降低到13%,而岩藻糖化丰度在AD中没有显著变化(图3A)。他们还观察到AD中复杂/杂化(既非岩藻糖化也非唾液酸化)聚糖修饰的丰度显著增加(图3A)。此外,定量分析显示,在AD患者大脑中,含有6个HexNAc残基的聚糖或>6个HexNAc残基的聚糖(高度分支或伸长的聚糖)修饰丰度显著降低,表明AD患者的N-聚糖分支和/或伸长减少(图3B)。
接下来,他们对164种不同糖组成的修饰进行了差异丰度分析,发现AD患者与对照组相比有显著改变的46种糖修饰,包括30种丰度降低的糖修饰和16种丰度增加的糖修饰(图3D)。基于糖修饰丰度谱的无监督层次聚类分析表明,鉴定出的46种糖修饰提供了区分AD患者和对照组的N-糖疾病特征(图3E)。在AD中,多糖聚类树状图显示了聚糖修饰在下调和上调聚类中的分离。
AD 中约 83% 的已鉴定下调的聚糖修饰是唾液酸化,主要是高度分支化和/或伸长的唾液酸化聚糖(例如 H8N6S1、H7N6F1S4 和 H8N7F1S3),而没有任何上调的聚糖修饰是唾液酸化(图3E),证明了唾液酸化和N-聚糖分支/延伸减少与AD之间的关联。 AD中鉴定的上调的聚糖修饰包括H4N2和H5N2聚糖的甘露糖基化、具有相对简单的中性结构的未修饰的复合/混合聚糖的修饰(例如H3N3、H5N3、H4N4和H5N4),以及通过平分含有GlcNAc的核心岩藻糖基化聚糖(例如H3N4F1、H3N5F1和H3N6F1)进行修饰。他们还观察到壳二糖核心型聚糖 N1 和 N2 以及寡甘露糖聚糖 H1N2、H2N2 和 H3N2 的修饰增加(图 3E),表明 AD 中 N-聚糖截短增加。

图3. AD 中 N-聚糖修饰总体水平与疾病相关的变化。
(A-C) AD 与对照大脑中指定聚糖类型、聚糖分支类别和前七种最丰富的聚糖成分的聚糖修饰的相对丰度的比较。(D) 差异倍数分析。(E) 基于聚糖修饰丰度图谱的无监督层次聚类分析。
04
AD 相关蛋白糖型具有改变的位点特异性 N-聚糖修饰
接下来,他们通过差异糖型丰度分析评估了各糖型的疾病相关丰度变化(图4A),并确定了与对照大脑相比,AD中水平发生显著改变的556个糖型,包括糖型丰度增加的149个糖蛋白中的421个糖型,以及糖型丰度降低的58个糖蛋白中的135个糖型(图4A-B)。糖型丰度变化与来自同一队列的脑样本中测定的相应蛋白丰度变化的比较表明,AD患者中观察到的大多数糖型丰度变化是由位点特异性N-糖修饰水平的变化引起,而不是由蛋白质表达水平的改变引起(图4C)。AD患者中449种糖型的位点特异性N-糖基化修饰水平是对照组的1.3倍,其中包括来自111种糖蛋白的337种糖型的N-糖基化修饰水平升高,以及来自52种糖蛋白的112种糖型的N-糖基化修饰水平降低(图4C)。无监督层次聚类分析显示,鉴定出的449种糖型可分为下调和上调两类,它们提供了区分AD患者和对照组的糖型特征(图4D)。在下调的聚类中,仅唾液酸化和唾液酸-岩藻糖化的聚糖富集;在上调的聚类中,仅岩藻糖化的聚糖、寡甘露糖型和寡甘露糖型聚糖富集(图4D)。与疾病相关糖型相连的N-糖基为AD提供了位点特异性糖基特征(图4D)。
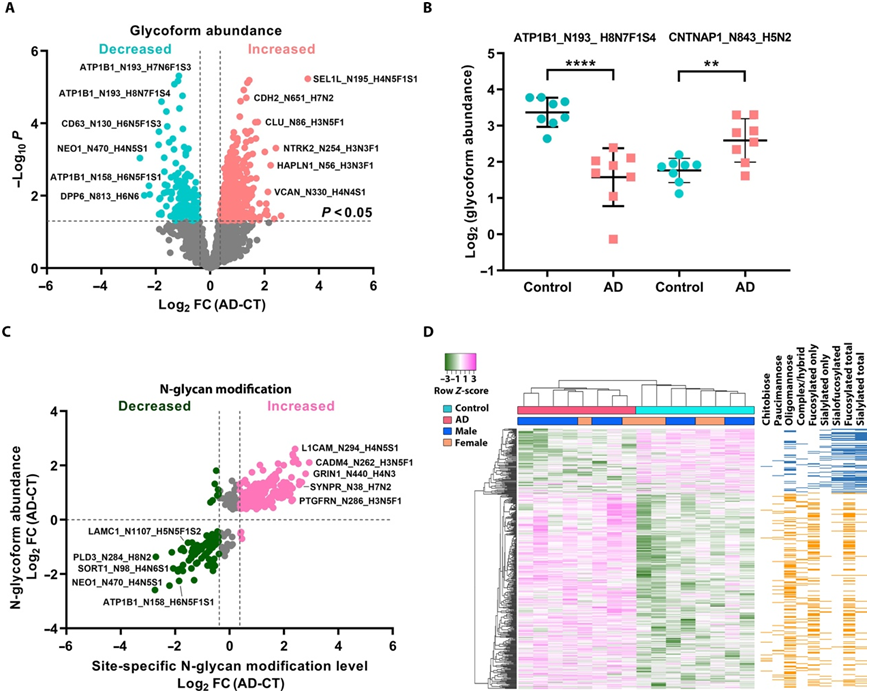
图4. 鉴定具有改变的位点特异性 N-聚糖修饰的 AD 相关蛋白糖型。
(A) AD 中单个糖型丰度的差异倍数。(B) AD 中丰度减少或增加的已识别糖型的示例。(C) 散点图显示大多数已识别的 AD 相关糖型的位点特异性聚糖修饰。(D) 基于已识别的 449 个具有改变的位点特异性聚糖修饰的糖型,对个体临床病例进行无监督的层次聚类。
除了糖基化修饰水平发生改变的糖型,他们还从105个糖蛋白中鉴定出179个具有位点特异性N-糖基化修饰的AD糖型,以及从32个糖蛋白中鉴定出49个位点特异性N-糖基化修饰的AD糖型(图5A-C)。总之,他们从178种高糖基化蛋白中鉴定出516种高糖基化糖型,这些高糖基化糖型在AD中增加或增加了位点特异性N-糖基化修饰;从71种低糖基化蛋白中鉴定出161种低糖基化糖型,这些低糖基化糖型在AD中减少或失去了位点特异性N-糖基化修饰(图5A-C)。高糖基化和低糖基化数据集都包含有寡甘露糖基化、寡甘露糖基化、岩藻糖化和/或唾液化改变的糖型和糖蛋白。他们对高糖基化和低糖基化数据集进行了GO富集分析,以了解AD中失调的N-糖基化修饰对细胞功能和过程的影响(图5D-F)。低糖基化和高糖基化数据集显著富集了参与细胞黏附、细胞表面受体信号传导、神经系统过程和细胞外基质(ECM)功能的细胞质膜和细胞外蛋白等相关通路。然而,只有AD相关的高糖基化与神经递质受体活性和信号素受体活性的通路显著相关,而AD中的低糖基化与内肽酶调节活性和钾离子转运的调节的通路显著相关(图5D-F)。本项研究揭示了AD中多种细胞功能受到不同类型糖修饰的不同影响。例如,在AD中,高寡聚甘露糖基化、低聚岩藻糖基化和低糖基化与ER和溶酶体显著相关,而其他类型的糖修饰变化与高尔基体无显著相关,而高寡甘露糖基化选择性地与高尔基体相关(图5D)。AD中的高寡甘露糖基化和高聚岩藻糖基化与Aβ结合特异性相关(图5E),并且AD中的高寡甘露糖基化和低糖基化优先与生物质量的调节相关(图5F)。
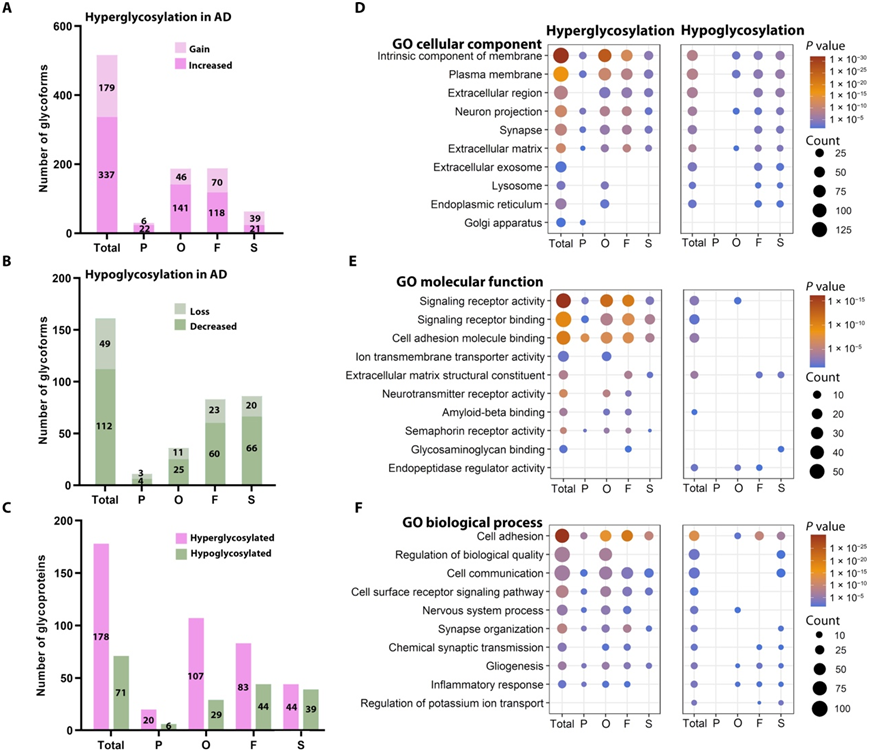
图5. AD 中改变位点特异性 N-聚糖修饰的糖型和糖蛋白的分析和注释。
(A-C) 高糖基化包括糖型和糖蛋白,在 AD 中具有增加的位点特异性 N-聚糖修饰和/或增加位点特异性 N-聚糖修饰,而低糖基化包括糖型和糖蛋白AD 中位点特异性 N-聚糖修饰减少和/或位点特异性 N-聚糖修饰完全丧失。(D-F)GO分析。
05
聚糖修饰共调节网络分析揭示了与 AD 病理学相关的聚糖模块
为了从系统水平深入了解人类大脑中的糖修饰及其在AD中的变化,他们利用加权相关网络分析(WGCNA)算法,基于164个糖组成的糖修饰丰度图谱的两两关联模式,构建了一个代表糖修饰的节点与连接由连接性定义的边相连的糖修饰协同调节网络(图6A)。通过应用无标度拓扑准则,他们生成了一个由8个强协同调控的糖修饰模块组成的人脑糖网络(图6A-B)。这些糖基模块(GNM)使用WGCNA约定的颜色编码,根据模块大小进行标记,从GNM1(最大的模块含52个糖基)到GNM8(最小的模块含5个糖基)(图6B)。他们为每个模块计算了一个特征图作为模块代表,并进行了模块-性状关联分析,以评估每个模块特征图与AD相关表型性状和脑样本其他特征之间的相关性。他们确定了1个正相关的聚糖模块(GNM2)和2个负相关的聚糖模块(GNM1和GNM4),它们与AD状态、aβ病理[阿尔茨海默病建立登记联盟(CERAD)评分]和/或神经原纤维缠结病理(Braak分期)显著相关,但与年龄、性别、ApoE基因型或死亡时间间隔无关(图6B)。与AD呈正相关的模块GNM2在AD中模块糖基化修饰水平显著升高,而与AD呈负相关的模块GNM1和GNM4在AD中模块糖基化修饰水平显著降低(图6C)。
与AD神经病理学(CERAD)强正相关的GNM2模块包含了25种糖基修饰(图6D),其中包括寡甘露糖基糖(H4N2、H5N2、H6N2和H7N2)、相对简单、中性结构的未装饰复合/杂交糖基(例如H3N3、H3N4和H5N4)、含有双重GlcNAc的核-岩藻糖基糖(例如H3N4F1、H3N5F1和H3N6F1)、低甘露糖基糖(H1N2、H2N2和H3N2)以及壳二糖核心糖基(N1和N2)。相比而言,与AD神经病理学呈强负相关的GNM1模块包含52个糖基修饰(图6D),主要包括唾液酸化-岩藻糖基化糖基(例如H5N6F1S2、H7N7F1S3和H8N7F1S3)、仅唾液酸化糖基(例如H6N5S3、H7N6S3和H7N6S4)以及高度分支和/或长形的糖基(例如H4N9、H10N10F1和H11N11S1)的修饰。他们发现核心岩藻糖的聚糖N1F1, N2F1、H1N2F1、H4N2F1和H5N2F1在AD负相关的GNM1模块中共分离,而它们的非核心岩藻糖的对应聚糖(N1、N2、H1N2、H4N2和H5N2)在AD正相关的GNM2模块中共分离(图6D),表明这些聚糖的核心岩藻糖降低与AD相关。与Aβ病理呈负相关但与tau纠结病理无相关性的GNM4模块含有7种糖修饰(图6D),包括由唾液酸化-岩藻糖基化的糖链H6N3F1S1和H9N6F1S2、仅唾液化的糖链H5N3S1和H8N6S1,以及高度分枝或长链的糖链H9N9F1所修饰,表明Aβ对这些糖链修饰具有特异性的负效应。
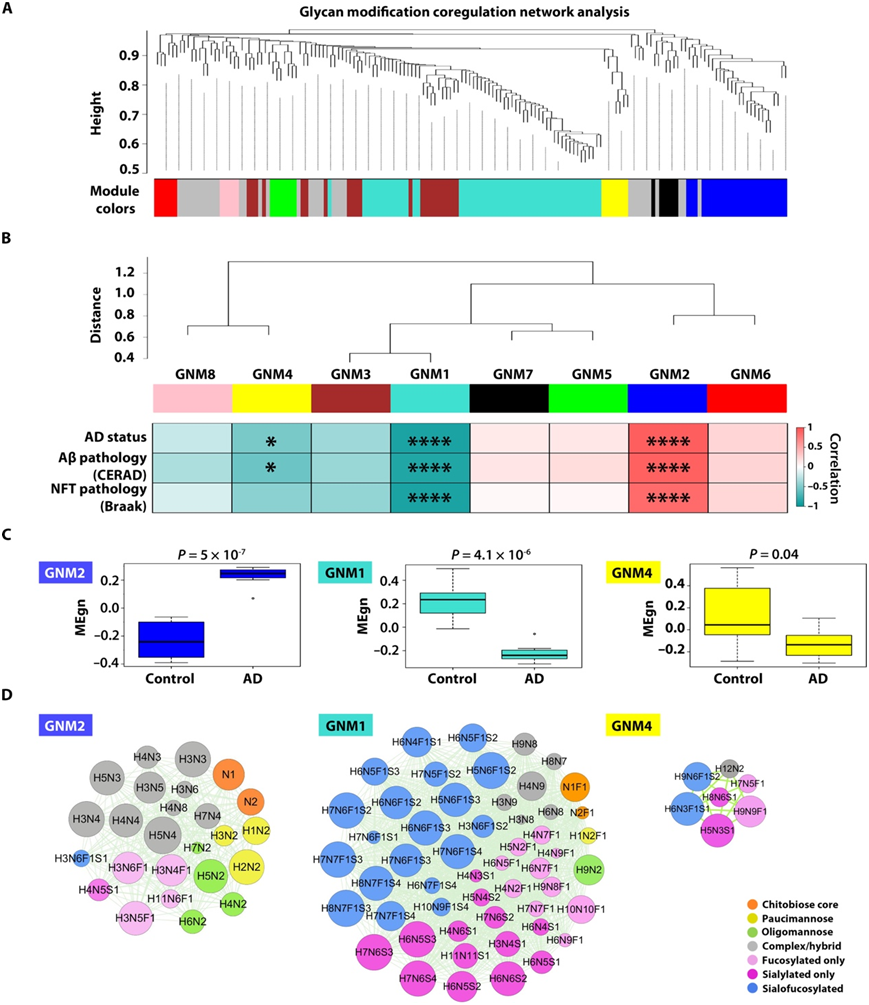
图6. AD相关聚糖模块的聚糖修饰共调控网络分析与鉴定。
(A) 聚糖 WGCNA 聚类树状图。 (B) 已识别的八个聚糖模块的模块间连接。(C) 每个 AD 相关模块的 AD 和对照案例中模块特征聚糖 (MEgn) 值的箱线图。(D) AD 相关模块 GNM2、GNM1 和 GNM4 的聚糖共调节网络图。
06
蛋白质糖型共调节网络分析揭示 AD 相关糖型模块
为了阐明 AD 中具有不同位点特异性聚糖修饰的蛋白质糖型之间的关系,他们使用 WGCNA 进行了糖型共调节网络分析,以构建一个糖型网络,其中的节点表示与基于糖型丰度谱的成对相关模式的连接定义的边缘连接的糖型(图7A)。网络分析揭示了人脑糖蛋白组组织成由 21 个共调节糖型模块组成的网络(图7A-B)。这些糖型模块(GFM)标记为GFM1(最大的模块包含159个糖蛋白中的440个糖蛋白)到GFM21(最小的模块包含16个糖蛋白中的36个糖蛋白)(图7B)。在糖型网络中,单个蛋白的不同糖型往往存在于不同模块中,如GABA-B受体亚单位GABBR1糖型对GFM2(GABBR1_N440_H5N2和GABBR1_N440_H3N5F1糖型)、GFM4(GABBR1_N482_H5N2糖型)、GFM8 (GABBR1_N440_H4N4F1糖型)、GFM14 (GABBR1_N502_H4N5S1糖型)和GFM21 (GABBR1_N440_H5N4F1糖型)模块的差异分布。一些糖型在同一模块中的共定位(例如GFM2 模块中存在两个 GABBR1 糖型)表明这些糖型及其位点特异性聚糖修饰的协调调节,而糖型划分为单独的模块(例如分布GFM2、GFM4、GFM8、GFM14 和 GFM21 模块中 GABBR1 糖型的差异)可能反映了位点特异性聚糖修饰的差异调节或不同糖型的不同细胞或亚细胞分布。
他们使用模块特征糖型作为模块代表进行模块-性状关联分析,并确定了与 AD 状态、Aβ CERAD 评分和/或 tau Braak 阶段显著相关的 7 个糖型模块,其中包括 5 个正相关模块(GFM1、GFM2、GFM4、GFM11和GFM19)和两个负相关模块(GFM3和GFM14)(图7B)。GFM1 和 GFM11 模块也与 ApoE4 等位基因剂量呈正相关(图 7B),这与 ApoE4 等位基因增加 AD 风险的剂量依赖性作用一致。此外,他们还发现了一个与 AD 表型无关的 ApoE4 正相关模块 (GFM6),表明 ApoE4 诱导的位点特异性聚糖修饰的一些变化与 AD 病理无关。所有五个AD正相关模块的模块糖型丰度谱在AD中显著增加(图7C),而两个AD负相关模块的模块糖型丰度谱在AD中显著降低(图7D)。
然后,他们评估了糖型模块中不同脑细胞的细胞类型标记物的富集情况,发现七个 AD 相关糖型模块中的 6 个(GFM1、GFM2、GFM3、GFM4、GFM11 和 GFM19)高度富集了神经元细胞标记物,但是其中一些模块也富含星形胶质细胞和/或少突胶质细胞标记物(图7B)。相比之下,AD相关的GFM14模块优先富集少突胶质细胞标记物(图7B)。使用模块本征糖型之间的成对相关性分析模块间关系揭示了紧密互连的糖型模块的高阶组织成超聚类(图7B)。 AD负相关的GFM3和GFM14模块在超聚类中的共定位(图7B)表明与这两个负相关的糖型模块相关的糖通路和/或生物过程之间的密切关系。五个 AD 正相关模块中的四个(GFM1、GFM4、GFM11 和 GFM19)共定位于另一个超聚类中(图 7B),表明这些正相关模块的相应糖通路和/或生物过程也可能在 AD 中具有功能相关和协调调节。
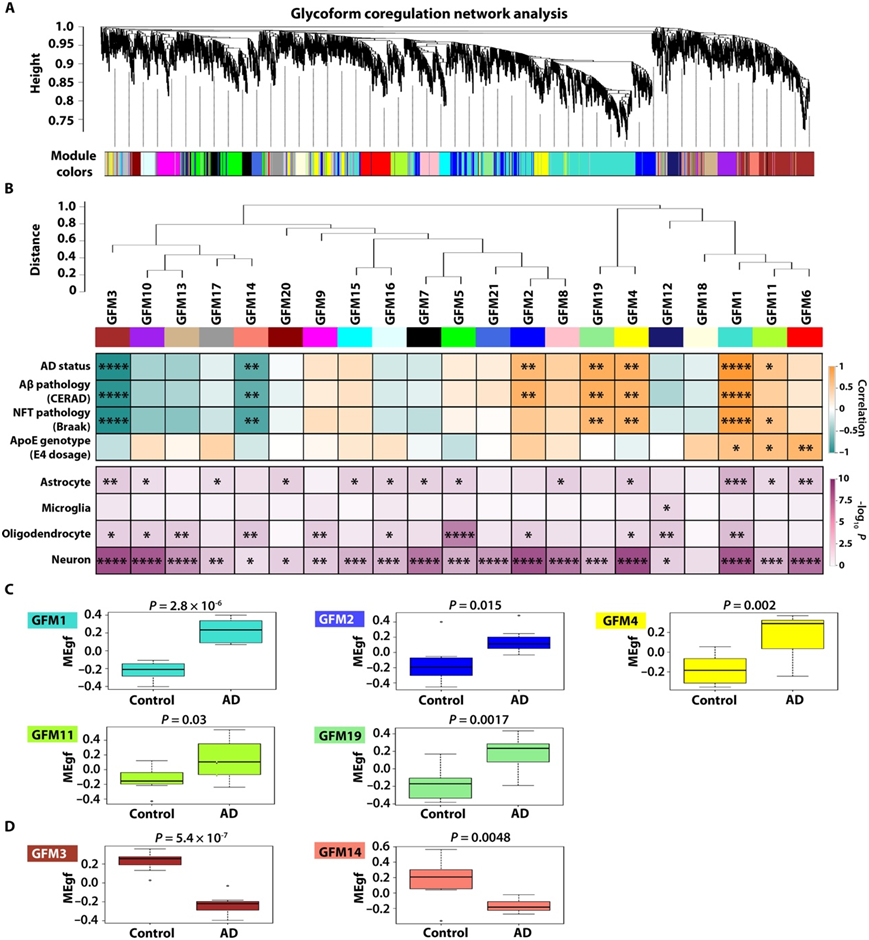
图7. 蛋白质糖型共调节网络分析和 AD 相关糖型模块的鉴定。
(A) 糖型 WGCNA 分析识别出 21 个共调节蛋白糖型模块。 (B) 21 个糖型共调节模块的模块间连接。(C-D) AD 正相关模块和 AD 负相关模块的 AD 和对照病例中模块表征糖型 (MEgf) 值的箱线图。
07
糖型模块分析揭示了 AD 中因聚糖修饰失调而改变的多个 CNS 过程
鉴于中心节点在确定网络模块的架构和功能中的核心作用,他们使用模块内连接性确定了每个模块的中心糖型,并在每个网络图的中心描绘了前 10 个最常连接的中心糖型(图8A)。通过分析每个模块中糖型上附着的位点特异性聚糖类型和聚糖组成的分布来评估每个 AD 相关模块的聚糖特征(图8B-C),发现每个 AD 相关模块都有独特的特征。位点特异性聚糖修饰模式,反映了糖型之间的模块特异性共调节及其在模块中的位点特异性聚糖修饰。例如,GFM1 模块富集了携带壳二糖核心、寡甘露糖、低聚甘露糖或仅岩藻糖基化聚糖的糖型,而对仅唾液化或唾液酸-岩藻糖基化聚糖的糖型的糖型缺乏代表性,而 GFM3 模块富集了唾液酸岩藻糖基化和仅唾液酸化的糖型(图8B)。寡甘露糖基化糖型的富集和唾液酸-岩藻糖基化糖型的耗尽是所有五个 AD 正相关模块共有的共同特征,而 AD 负相关模块的情况恰恰相反,其唾液酸-岩藻糖基化糖型富集,而寡甘露糖基化糖型耗尽(图8B)。
他们随后进行了GO富集分析,发现所鉴定的AD相关糖型模块具有重叠但不同的生化和功能特征(图8D-F)。所有7个与疾病相关的模块均与细胞黏附过程显著相关,且细胞黏附蛋白的糖型过度表达,包括神经细胞黏附分子(例如NCAM1、NCAM2、NRCAM、L1CAM和CHL1/L1CAM2)、突触细胞黏附分子(CADM1/SynCAM和CADM2/SynCAM2)、细胞黏附分子的IgLON家族(OPCML、LSAMP和NEGR1)、黏附G蛋白偶联受体(ADGRB1/BAI1、ADGRB3/BAI3和ADGRL1/latrophilin-1)、neurexin1 (NRXN1)、神经神经蛋白-4 (TENM4)、神经元正五聚蛋白-1 (NPTX1)、神经元正五聚蛋白受体(NPTXR)、接触蛋白(CNTN1、CNTN2、CNTN3和CNTN4)、接触蛋白相关蛋白(CNTNAP1/Caspr1、CNTNAP2/Caspr2、CNTNAP4/Caspr4和CNTNAP5/Caspr5)、钙黏蛋白(CDH2、CDH10和CDH13)、原钙黏蛋白(PCDH1、PCDH9和PCDH17)、神经束蛋白(NFASC)、新生成素(neogenin)、神经活酶蛋白(NPTN)、ADAM22、LGI1、CD47、CD200、SIRPA、JAM3和/或THY1(图8E-F)。从细胞黏附蛋白中鉴定出的位点特异性糖型是AD正相关模块(如L1CAM_N671_H3N4F1, NRCAM_223_H4N2, CNTN1_N338_H5N2和OPCML_N70_H6N2)和AD负相关模块(CD200_N90_H6N6F1S2和THY1_N42_H5N4F1糖型)的核心糖型,支持细胞黏附糖型作为AD疾病相关糖型网络改变的关键驱动因素的核心作用。
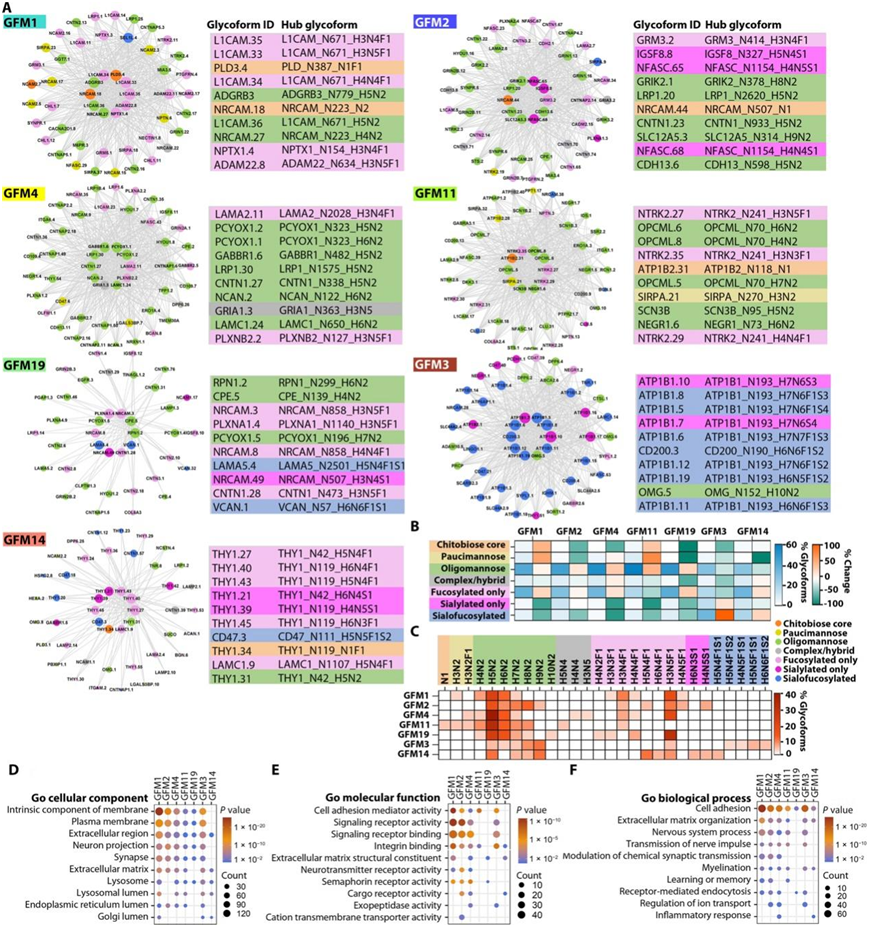
图8. AD 相关糖型模块网络的生化和功能特征。
(A) AD 相关模块的糖型共调节网络图,节点代表单个糖型。(B) 热图显示每个 AD 相关模块中具有指定聚糖类型的糖型百分比。 (C) 热图显示了每个 AD 相关模块中具有前 10 个最常见聚糖成分的糖型的百分比。 (D-F) GO分析。
08
AD 中 N-聚糖修饰酶和调节因子的变化
为了确定N-糖基化酶和调节因子的改变是否参与驱动AD脑内观察到的N-糖基化修饰变化,他们利用已发表的AD转录组Meta数据,分析了来自宗教教团研究(Religious order Study)和记忆与衰老项目(Memory and Aging Project, ROSMAP)、西奈山医学院(Mount Sinai School of Medicine, MSSM)以及梅奥研究的转录组数据集中N-糖基化相关基因的mRNA表达谱(图9)。此外,他们在来自同一队列的蛋白质组数据和其他5个AD脑蛋白质组数据集中评估了N-糖基化酶和调节因子的蛋白质表达模式(图9)。OST酶复合物的几个亚基(如STT3A、STT3B、RPN2、TUSC3和MAGT1)的表达水平在AD大脑中发生了改变(图9),这与之前确定的OST失调是AD中蛋白质N-糖基化位点占据变化的原因一致,OST酶复合物通过共翻译或翻译后将预先形成的寡糖Glc3Man9GlcNAc2转移到ER中蛋白质的天冬酰胺残基来催化N-糖基基化的起始步骤(图 9)。
多组学研究表明,葡萄糖苷酶I (MOGS)、葡萄糖苷酶II的GANAB和PRKCSH亚基(这些酶从糖蛋白上的Glc3Man9GlcNac2聚糖上依次剪切三个末端葡萄糖残基)改变了AD脑内的蛋白质水平(图9)。此外,他们发现内质网凝集素malectin (MLEC)和内质网凝集素伴侣calnexin (CANX)和calreticulin (CALR)与内质网凝集素伴侣MLEC(MLEC与内质网糖苷酶I生成的二糖基化糖结合以保留内质网中未成熟或错误折叠的蛋白)以及尿苷二葡萄糖:糖蛋白葡糖基转移酶UGGT1和UGGT2。蛋白质折叠传感器将未完全折叠的蛋白质重新葡糖基化,从而与CANX-CALR伴侣系统重新结合以进行进一步折叠,在AD大脑中也发生了改变(图9)。
他们还发现了与认知功能(MMSE30评分)、Aβ病理(CERAD评分)和/或NFT病理(Braak分期)显著相关的其他N-糖基化相关蛋白(STT3A、DAD1、TUSC3、UGGT2、MAN2A2、LMAN2L、OS9、ERLEC1、FUT9、HEXB和MANBA)(图9)。此外,对Aβ斑块和NFT蛋白质组学数据的研究显示,一些N-聚糖修饰酶(RPN1、DDOST、GANAB、PRKCSH、UGGT1和HEXB)和ER凝集素伴侣蛋白(CALR和CANX)在AD脑内与Aβ斑块和NFT共定位(图9)。
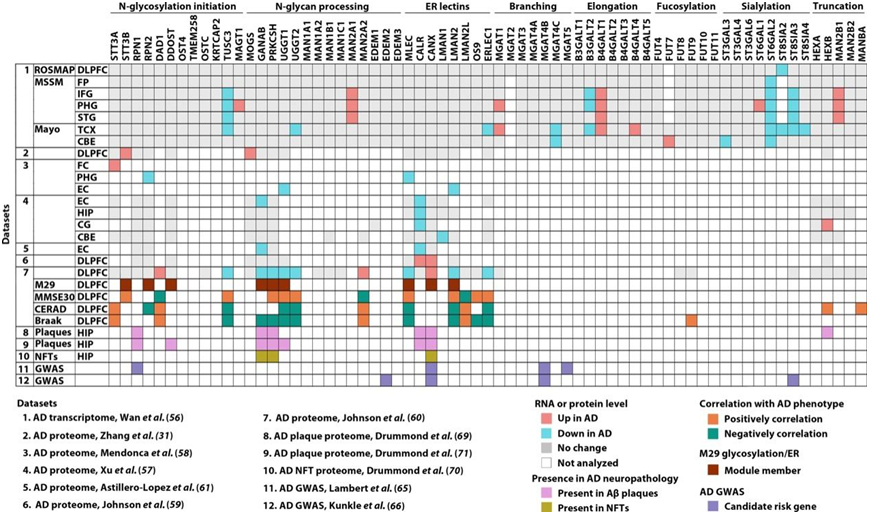
图9. 多组学研究揭示了 AD 中 N-糖基化机制的改变
+ + + + + + + + + + +
结 论
本项研究使用基于完整糖肽的定量糖蛋白组学与系统生物学相结合,对人类 AD 和对照大脑进行了全蛋白质组糖型分析研究。从近 1200 种糖蛋白中鉴定出超过 10000 种人脑 N-糖型,并发现了糖型和聚糖修饰改变的疾病特征,包括 AD 中唾液酸化和 N-聚糖分支和延伸的减少以及甘露糖基化和 N-聚糖截的增加。网络分析揭示了脑糖蛋白组的高阶组织为共同调节的糖型和聚糖网络,并发现了与 AD 临床表型、β-淀粉样蛋白积累和 tau 病理学相关的糖型和聚糖模块。本项研究结果提供了关于疾病发病机制的宝贵见解以及 AD 中糖型和聚糖变化的丰富数据,并为开发基于糖基化的 AD 疗法和生物标志物提供了新的理论依据。
+ + + + +



