

 English
English文献解读|MICROBIOME(16.837):马铃薯赤霉病的发生与土壤微生物群落的组成和功能有关
✦ +
+
论文ID
原名:The occurrence of potato common scab correlates with the community composition and function of the geocaulosphere soil microbiome
译名:马铃薯赤霉病的发生与土壤微生物群落的组成和功能有关
期刊:Microbiome
影响因子:16.837
发表时间:2019.02.01
DOI号:10.1186/s40168-019-0629-2
背 景
土壤微生物可以介导植物病害的发生。马铃薯赤霉病(CS)是一种由致病性链霉菌Streptomyces引起的世界性、难治性疾病,但目前对CS与土壤微生物之间的相互作用知之甚少。这项研究分析了具有高(H)和低(L)疮痂严重程度的马铃薯植物的四个土壤-根系系统区间:(地层土壤(GS)、根瘤土壤(RS)、根区土壤(ZS)和沟渠土壤(FS)),检查了疮痂毒素生物合成基因txtAB和细菌16S rRNA基因的拷贝数以及四个土壤系统区间的多样性和组成。
在土壤-根系系统中,GS微生物群与CS的严重程度密切相关。CS的发生伴随着群落组成和功能的变化。这些差异功能为阐明CS发生与土壤微生物群之间相互作用的机制提供了新线索,而不同的群落组成为CS发生提供了新的见解。
实验设计
结 果
01

低疮痂的特征是GS微生物群txtAB基因丰度低、细菌丰度低和细菌多样性高
对ZS和FS的理化特性进行了检测,分析结果表明,CS发生在酸性土壤条件下,在H组和L组之间,ZS中铵(NH4+-N)的浓度存在显著差异,在FS中观察到的所有理化特性没有显著差异。除铵外,H组和L组之间的土壤总碳TC和有机物OM浓度也存在显著差异。
txtAB是CS毒素thaxtomin生物合成的关键基因,能够准确定量致病链霉菌菌株。为了确定致病链霉菌的丰度,检测了所有土壤样本的txtAB基因的拷贝数,只有GS在qPCR中产生清晰的荧光信号(图1a),这表明GS含有大量的致病链霉菌。此外,GSH和GSL的txtAB拷贝数存在显著差异,且与CS严重程度呈显著正相关。
通过qPCR检测细菌16S rRNA基因拷贝数,与GS相比,RS的细菌拷贝数更高,并且GSH和GSL之间只有GS的细菌拷贝数存在显著差异(图1b)。GS的细菌拷贝数与疮痂严重程度呈显著正相关,表明GS中细菌丰度的增加可能是CS发生的信号。
所有土壤样本的进行16S rRNA测序,进行OTU分析。从细菌群落中共产生了2700个OTU,GSH中的OTU数量低于GSL(图1c)。GS的细菌OTU数与疮痂严重程度呈显著负相关。

图1 GS的细菌群落
a txtAB基因拷贝数 b细菌16S rRNA基因拷贝数 c OTU数量
02
高疮痂和低疮痂GS的细菌群落组成存在显著差异
对细菌OTU使用Bray-Curtis度量进行主坐标分析(PCoA)。所有的样品按土壤区划类型聚类(图1d)。在同一土壤根系分区内,GS中的细菌群落可以根据CS进行区分(图1e),但RS、ZS、FS的情况并非如此(图1f、g、h)。这表明,GSH和GSL细菌群落组成存在显著差异。
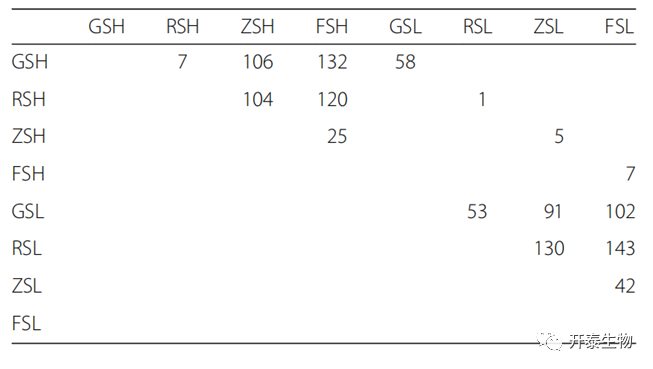
各组之间属级细菌相对丰度的差异与PCoA结果一致(表1)。在GSL和RSL之间发现了53个不同的细菌属,而在GSH和RSH之间只发现了7个不同的菌属,这表明H可能会减少GS和RS之间的群落差异。GS在H和L之间有58个分化的细菌属,大于RS、ZS和FS中分别发现的1、5和7个。进一步表明,GS的细菌群落组成与CS的严重程度有关。
表1 通过双尾Wilcoxon检验评估的组间显著差异的细菌属的数量
03
低疮痂的特征是GS微生物群致病性链霉菌丰度低
选择GS DNA进行鸟枪元基因组测序,获得了总共657254147个高质量的序列片段。与NR数据库对齐进行分类注释。分配给细菌的序列片段占据了主导地位(约98.03%),古细菌约占0.07%、真核生物约占1.44%、病毒约占0.41%。其中243560个序列片段代表的18个链霉菌,可能是CS病原体。S. acidiscabies是最主要的菌种,其次是S. niveiscabiei、S. turgidiscabies和S. scabiei.
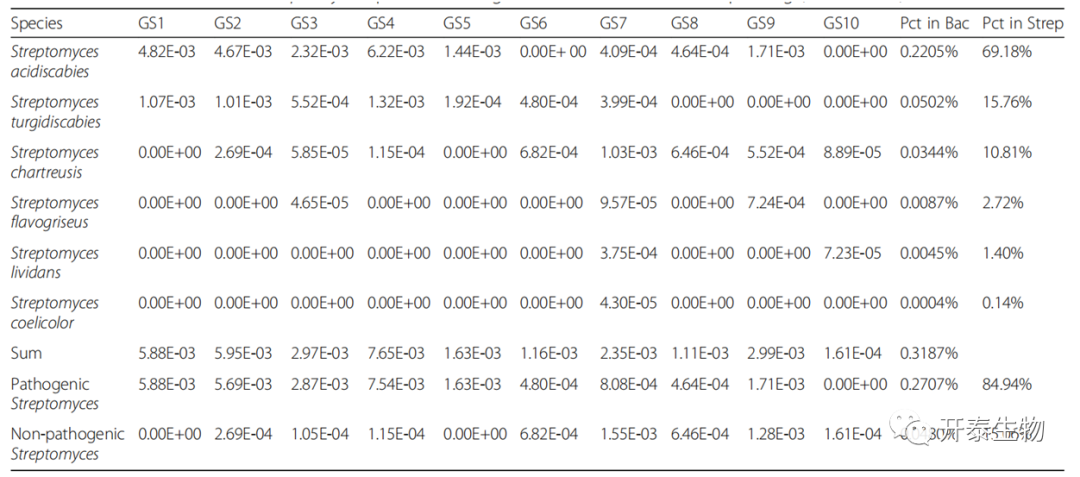
通过MetaPhlAn v.2.0软件量化微生物群落内的相对丰度。微生物群落的主要组成部分细菌被分离出来,S. acidiscabies 和S. turgidiscabies是可能的CS病原体(表2)。其中S. acidiscabies是样品中MetaPhlAn2分析的主要CS病菌,与NR排列结果一致。
在培养实验中,根据NCBI BLAST结果,11个分离菌株与S. acidiscabies菌株具有最高的系统发育相似性,1个与S. turgidiscabies菌株具有最高的相似性。进一步证实了S. acidiscabies和S. turgidiscabies的存在,并确定S. acidiscabies是本研究中的优势链霉菌种。S.acidiscabies和S. turgidiscabies的相对丰度之和为致病链霉菌的相对丰度。其他链霉菌种为非致病性(表2)。
表2 元基因组细菌中链霉菌的相对丰度
通过计算细菌的EAA发现,与GSL相比,GSH的致病链霉菌显著富集。致病链霉菌的EAA与txtAB基因拷贝数和疮痂严重程度表现出极其显著的正相关关系。致病链霉菌的丰度可以反映CS的严重程度,并且致病链霉菌的EAA比相对丰度更适合反映txtAB基因拷贝数和CS严重程度。
04
与CS相关的GS微生物群的群落组成和功能
根据元基因组测序数据,细菌群落由116个属组成。选择疮痂严重程度、致病链霉菌的EAA和txtAB基因拷贝数三个参数,使用Spearman相关分析法对细菌属的EAA进行分析(图2)。

图2 元基因组细菌的EAA(属级)与疮痂严重程度、致病链霉菌的EAA和txtAB基因拷贝数之间的相互作用网络。
结果表明,28个属与CS严重程度显著相关。16个属与致病性链霉菌的EAA显著相关。27个属与txtAB基因拷贝数显著相关。共有13个属与所有3个参数均呈显著正相关。在所有相关属中,Variovorax表现出最高的EAA;Stenotrophomonas, Agrobacterium, Sphingobium, Streptomyces与3个参数显著正相关;Geobacillus、Curtobacterium和未分类的Geodermatophilaceae与3个参数显著负相关。
利用元基因组细菌群落的共现网络比较GSH和GSL之间的共现关系和群落复杂性。平均邻居数和网络密度均为GSL更高,说明与GSH相比,这些细菌在GSL中的共性关系更复杂。可能是GSL中更复杂的共生群落关系导致GSL群落对病原入侵更具抵抗力。
在微生物群落组成和功能方面,GSH和GSL之间的差异显著,表明GSH和GSL之间群落组成和功能都有差异(图3)。群落功能的差异系数值低于群落组成的差异系数,表明GS的微生物功能比其群落组成更相似。总的来说,疮痂严重程度与微生物功能的关系更为密切,且L的功能相似性高于H。
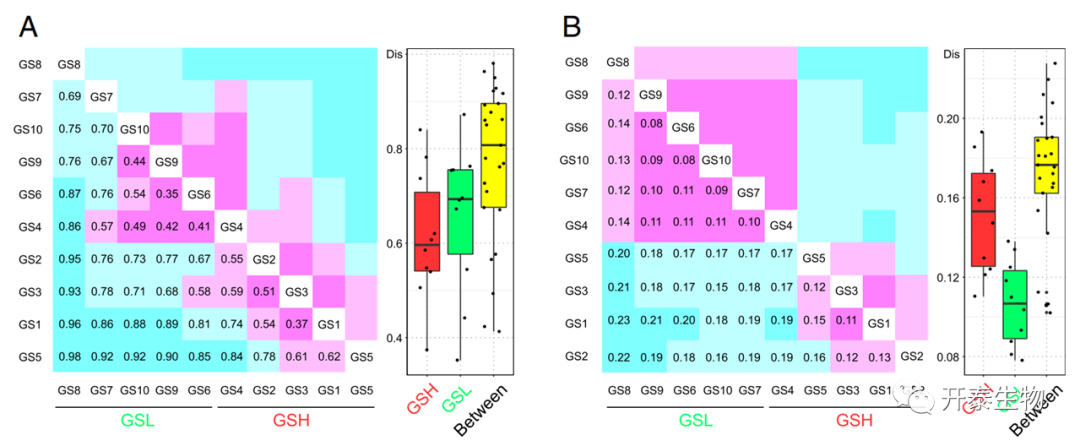
图3 A是元基因组细菌群落组成的EAA(属级)B是基于Bray-Curtis的KO功能类别的相对丰度的差异性
在GSH和GSL中分别发现了240和561个显著富集的KO功能类别(图4a)。保留了98个相对丰度大于0.03%的差异性KO功能类别(图4b),其中16个显著富集GSH,82个显著富集于GSL。在GSH和GSL中,"新陈代谢 "途径是主要的组成部分,但在GSL中有多条途径属于 "遗传信息处理",而在GSH中没有这样的途径。
为了进一步探索所有不同KO功能类别参与的途径,分别分析富集在GSH或GSL的KO功能类型的KEGG途径富集(图4c)。“氮代谢”途径显著富集在GSH。被认为参与硝酸盐和亚硝酸盐的运输以及一氧化氮的还原和甲酰胺的水解的基因和酶在GSH微生物群中的相对丰度也显著增加。参与 "药物代谢-细胞色素P450 "和 "细胞色素P450对异种生物的代谢 "的途径也富集在GSH,并可能在thaxtomin 植物毒素的生物合成中发挥重要作用。
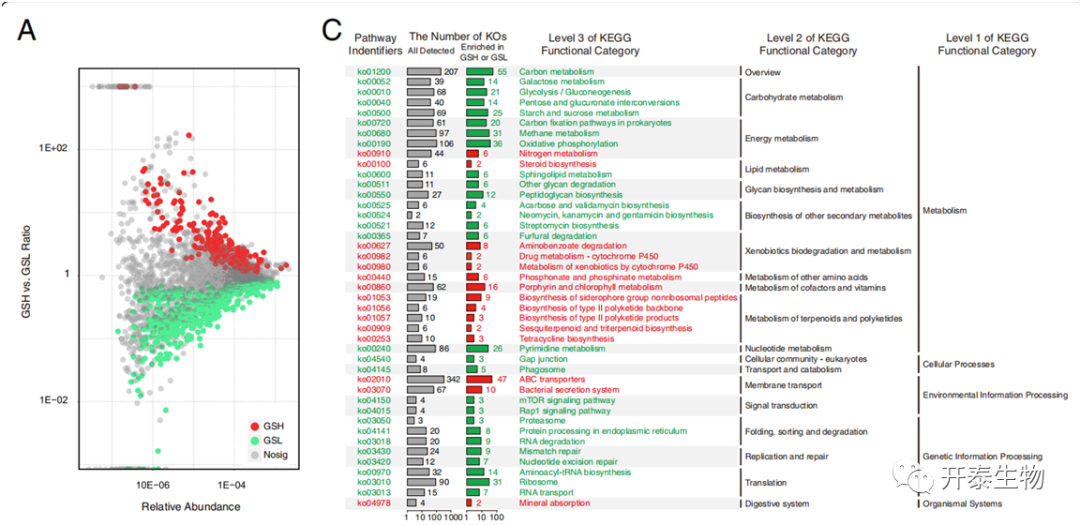
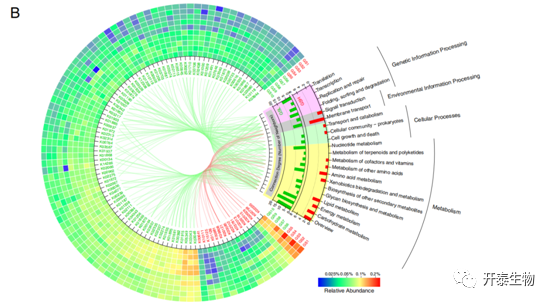
图4 GSH和GSL之间显著不同的KO功能类别和途径
(a)所有KO功能类别(b)KO功能类别(相对丰度>0.03%)(c)所有明显富集的途径
+ + + + + + + + + + +
结 论
本研究旨在探讨土壤微生物群与CS之间的相互作用,并证实GS的微生物群落组成和功能与CS严重程度相关。低txtAB基因丰度、低细菌丰度、高细菌多样性、高共现网络复杂性和高群落功能相似性是马铃薯CS严重程度低的指标。
与疮痂严重程度高的马铃薯相关的GS微生物组包含致病性相关基因图谱,H组显著富集涉及“ABC转运体”、“细菌分泌系统”、“QS”、“氮代谢”途径的几个基因以及细胞色素P450的一些代谢。相比之下,L组中某些抗生素的生物合成途径显著丰富。
研究拓宽了对CS发生与土壤微生物群之间关系的理解,并为CS发生提供了新的见解。
+ + + + +



