

 English
English文献解读|Hepatology(12.9):肝细胞癌的综合泛素化蛋白组学表征
✦ +
+
论文ID
原名:Integrated ubiquitomics characterization of hepatocellular carcinomas
译名:肝细胞癌的综合泛素化蛋白组学表征
期刊:Hepatology
影响因子:12.9
发表时间:2024.09.30
DOI号:10.1097/HEP.0000000000001096
背 景
患有侵袭性肝细胞癌(HCC)的患者的治疗选择有限。因此,需要更好地了解 HCC 的发病机制以改善治疗。HCC 的基因组研究提高了大众对癌症生物学的理解。然而,HCC 的泛素特征仍然知之甚少。
实验设计
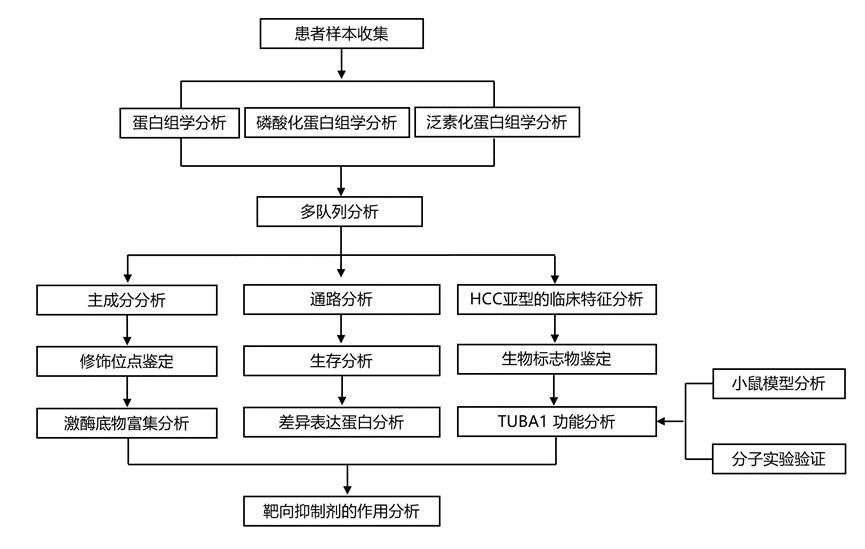
结 果
01

HCC样本的多组学研究
研究团队从未接受过化疗或放疗的患者中共收集了85例HCC肿瘤组织和18例癌旁正常组织(ANT)(图1A)。蛋白质组学分析采用基于质谱的非标记定量策略,共鉴定出6277个蛋白质。使用diGly修饰的肽(也称为K-ε-GG肽)富集策略进行泛素化蛋白组学分析,在5978个蛋白质上鉴定出30032个泛素化位点。他们对18对HCC进行了磷酸化蛋白质组学分析,确定了6002蛋白上的23558个磷酸位点。他们对来自85个肿瘤和18个ANT的蛋白质组学(3203个蛋白质)和泛素化蛋白组学(3099个泛素化位点)数据进行了主成分分析,数据显示了肿瘤和ANT之间的明确分离(图1B-C)。此外,他们还分析了18个ANT(左)和85个肿瘤(右)中每个蛋白质的泛素化位点数量。平均而言,每个肿瘤肝组织样本鉴定出3353个泛素化蛋白,每个ANT样本鉴定出2335个泛素化蛋白(图1D),表明肿瘤样本的泛素化位点多于ANT样本。此外,对4750个磷酸化修饰位点进行的主成分分析显示,18对肿瘤和ANT显著分离(图1E),共有23558个磷酸化修饰位点对应6002个磷酸化修饰蛋白(图1F)。
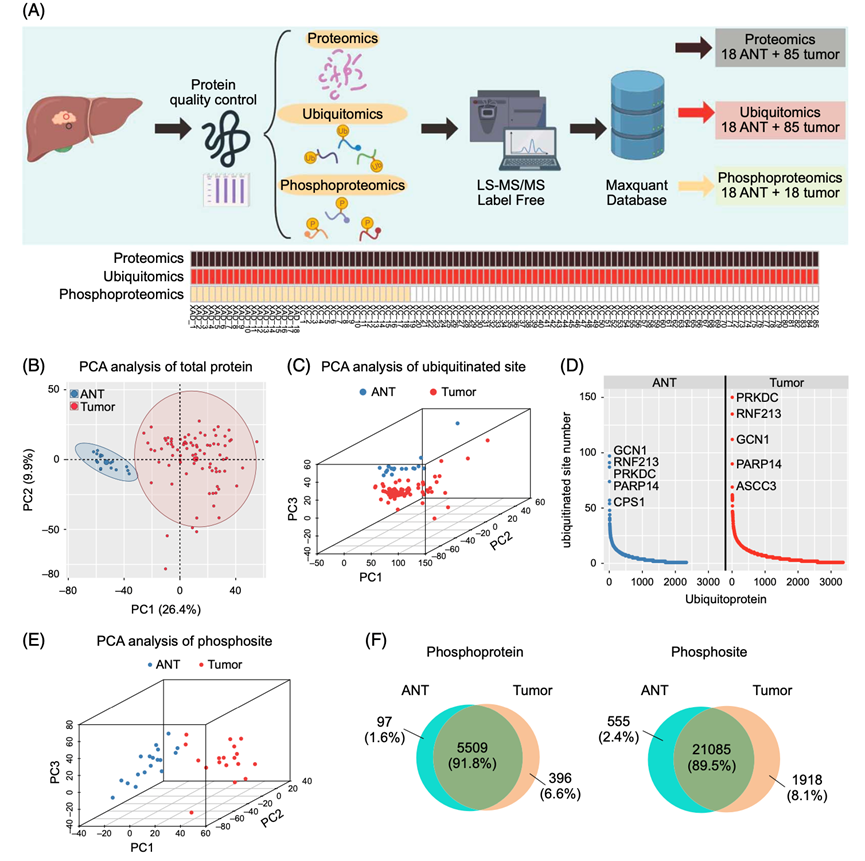
图1. HCC样本的多组学研究。
(A) HCC多组学分析示意图。(B) 85例HCC样本和18例ANT中3203个蛋白质的PCA。(C) 85例肝癌样本和18例ANT 中的3099个泛素化位点的PCA分析。(D) 肿瘤和ANT中泛素化蛋白中泛素化位点的分布。(E) 18对配对样本中有4750个磷酸位点。(F) 肝细胞癌患者的磷酸化蛋白质组学概况。
02
HCC潜在药物靶点的鉴定和磷酸化蛋白质组学和泛素化蛋白组学特征
蛋白质磷酸化是蛋白质最丰富的翻译后修饰,是细胞功能的关键调节因子磷酸化与泛素化修饰密切相关,广泛影响肿瘤的发生和其他多种疾病。在18对配对的肿瘤和ANT中,他们确定了4750个磷酸位点,其中与ANT相比,肿瘤中644个磷酸化修饰位点(13.56%)显著上调(红点),526个磷酸化修饰位点(11.07%)显著下调(蓝点)(图2A)。在这些磷酸化修饰蛋白中,与ANTs中的磷酸化修饰蛋白相比,在肿瘤中,149个(10.58%,红点)和97个(6.89%,蓝点)的磷酸化修饰蛋白分别出现(超过2倍的)增加和减少(图2B)。有趣的是,1170个磷酸位点发生了显著变化,包括1048个丝氨酸、115个苏氨酸和7个酪氨酸,这表明丝氨酸和苏氨酸磷酸化可能与肝癌发生相关(图2C)。热图显示了18对HCC肿瘤中下调或上调的前10种磷酸化修饰蛋白,ANT显示了已知在HCC中发挥肿瘤支持作用的蛋白质,如DKC1、LMNB1和CAD(图2D)。此外,p-CAD Ser1406与其蛋白水平和HCC患者低生存率呈负相关,p-LMNB1 Ser393与患者低生存率呈正相关,而p-DKC1 Ser451与其蛋白水平和HCC患者生存率无显著相关。对肿瘤和ANT之间的磷酸化蛋白质组学进行的激酶底物富集分析显示,肿瘤中有多种激酶(激酶标准化富集评分≥2)发生激活(激酶标准化富集评分≥2)(图2E),包括细胞周期蛋白依赖性激酶(CDK)家族成员(如CDK1、CDK5)、CDK样蛋白(CDKL)蛋白(CDKL1、CDKL4)、丝氨酸/苏氨酸蛋白激酶(FRAP、ICK)、丝裂原活化蛋白激酶(P38A、P38D)、双特异性蛋白激酶CLK1、SRSF蛋白激酶MSSK1和酪蛋白激酶(CK2A2)。
利用机器学习方法(L1LOG和L1SVM)训练从多源数据(药物子结构、药物靶标蛋白、靶标蛋白结构域、药物不良反应和药物治疗作用)获取的药物相关异构网络,建立和量化药物子结构-结构域关联关系,鉴定出排名前12位的癌症相关磷酸化蛋白及其候选药物(图2F)。值得注意的是,fostemsavir(一种潜在的靶向gp160的药物)、venetoclax(靶向Bcl-2的药物)和tivozanib(靶向VEGFR-1/2/3的药物)已由美国食品药品监督管理局(FDA)批准,可能成为治疗HCC的新药物。在本研究HCC队列中,CBR1 Ser151显示出高预后风险评分,并与不良预后呈正相关。
考虑到磷酸化和泛素化修饰之间的交互作用在控制蛋白质功能中的潜在作用,他们通过热图检测了HCC肿瘤和ANT的磷酸化蛋白质组和泛素化蛋白组特征。参与代谢的肿瘤富集蛋白主要表现为泛素化和磷酸化增加,而参与转移的肿瘤富集蛋白主要表现为高泛素化和低磷酸化修饰。值得注意的是,与肿瘤中的免疫富集相关的蛋白具有低泛素化和高磷酸化修饰(图2G)。这些分析表明,磷酸化蛋白质组和泛素化蛋白组数据具有独特的特征,当整合时,可以提供新的见解和机会,以鉴定新的功能重要的靶点。
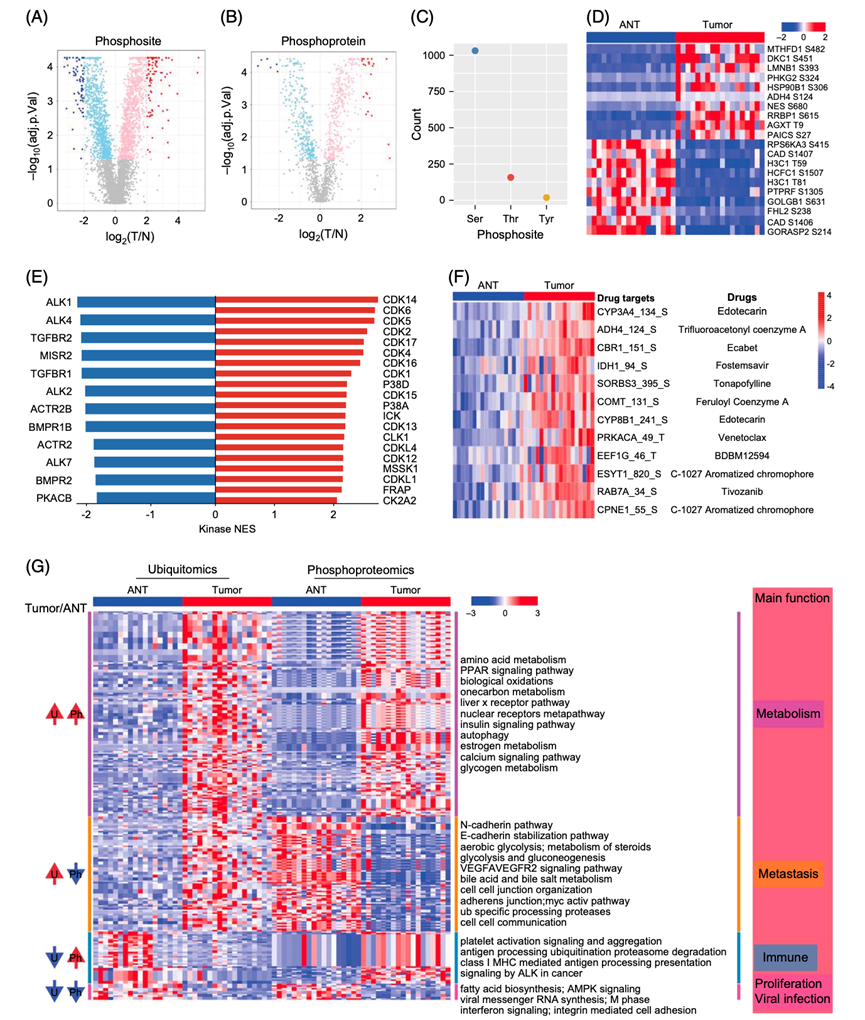
图2. 肝癌中蛋白磷酸化与蛋白泛素化或潜在药物靶点的相关性。
(A-B) 显示肿瘤和ANT中磷酸位点和磷酸化修饰蛋白表达的火山图。(C) 点图显示丝氨酸、苏氨酸和酪氨酸的丰度。(D) 热图显示18对HCC组织中表达最高的10个肿瘤/ANT和ANT/肿瘤的磷酸化位点。(E) 在肿瘤和ANT中通过激酶底物富集分析评价激酶活性。(F) HCC患者的潜在药物靶点。(G) 肿瘤和ANT中的差异表达蛋白和泛素化蛋白及其相关的生物学途径。
03
预后不良和伴/不伴HBV的HCC患者的蛋白质组学特征
HCC患者的临床生存仍然很差,世界卫生组织的预测强调了改善预后不良患者的结局的必要性。因此,通过蛋白质组学研究临床结局的差异可以提供新的见解。将无病生存(DFS)短(<所有患者DFS的1 / 4分)的患者定义为DFS-poor组,将DFS长(> 3 / 4分)的患者定义为DFS-good组。所有DFS-poor的患者均出现复发,而DFS好的患者均未出现复发(图3A)。基因集富集分析显示,生物氧化、脂肪酸代谢等代谢相关通路在DFS-good组富集。相反,与转移和增殖相关的通路,如黏着斑、细胞外基质(ECM)组织和细胞周期,在DFS-poor组中富集,表明激活的癌症相关信号通路与不良结局相关(图3B-C)。参与生物氧化的MAOA、含黄素的单氧合酶-3和细胞色素P450 8B1在DFS-good组中高表达,与良好预后呈正相关,而参与黏着斑的COL4A1,LAMC1和LAMA4,在DFS-poor组中高表达,与良好预后呈负相关(图3D)。
HBV感染是HCC的主要危险因素,占全世界HCC病例的一半以上。近年来的多组学数据集研究揭示了HBV相关HCC新的信号通路和潜在的预后生物标志物或治疗靶点,但非hbv相关机制仍有待探索。利用蛋白质组学数据进行基因集富集分析发现,hbv相关组的基因富集于胆固醇代谢和脂质代谢(图3E-F)。标志物磷酸果糖激酶肝型在hbv相关组中富集,并与HBV阳性患者的不良预后呈正相关(图3G)。相反,与氧化磷酸化和ECM重塑相关的通路,如ECM蛋白聚糖和ECM糖蛋白,在非HBV相关组中富集(图3E-F)。COX6B1,一种参与氧化磷酸化的蛋白,在HBV阴性的HCC肿瘤中高表达,并且在无HBV和HCC患者中与不良预后呈正相关(图3G)。总体而言,这些结果表明,预后不良的HCC患者存在信号通路多样性。
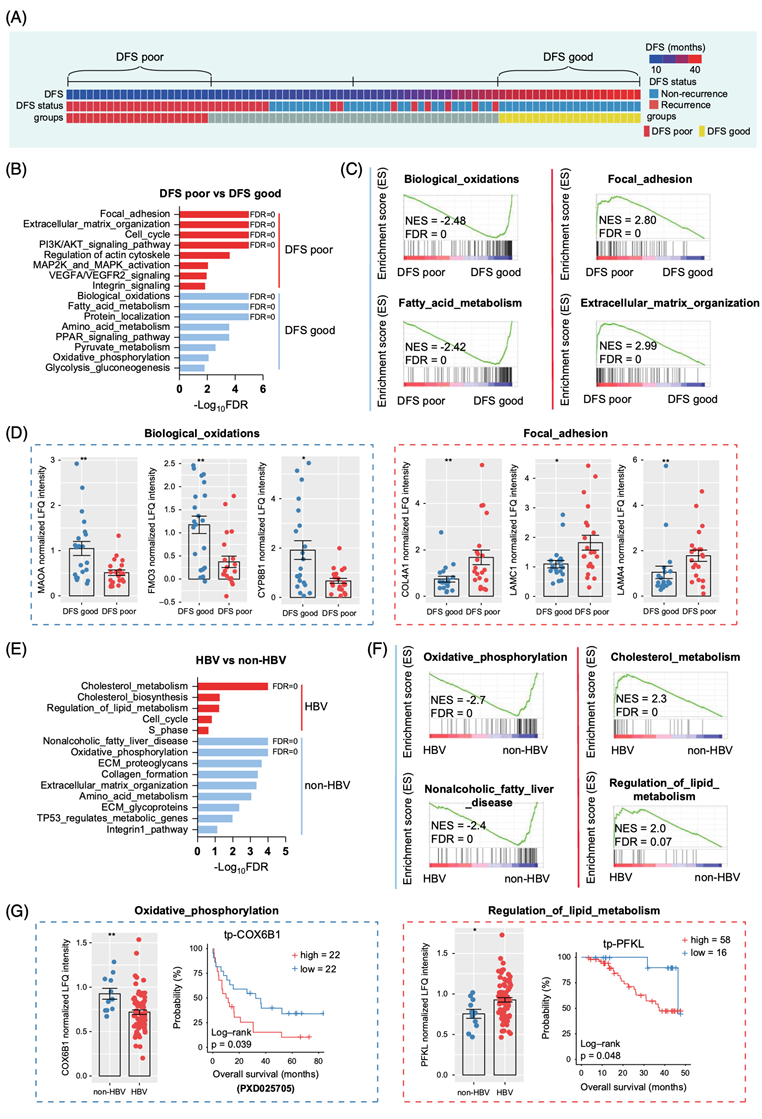
图3. HCC预后不良的蛋白质组学特征。
(A) 临床热图显示基于DFS的分组信息。(B-C) 蛋白质组学数据的GSEA揭示了与DFS良好或不良相关的通路。(D) 蛋白组学分析预后良好组和预后不良组之间的蛋白表达差异。(E-F)通路分析。(G)差异表达蛋白分析。
04
肝细胞癌的分子分型及其与临床预后的关系
基因组、转录组和蛋白质组信息已用于将HCC分为亚组。泛素化蛋白组学和蛋白质组学数据可能为HCC患者寻找潜在的诊断生物标志物或治疗靶点提供新的见解和更多的机会。然后,他们基于蛋白质组学和泛素化蛋白组学进行无监督聚类,在85个肿瘤中鉴定出3个具有不同临床特征的亚组(S-I、S-II和S-III)(图4A)。
S-I的特征是代谢相关蛋白水平最高,如FBP1, PKLR, GSTZ1和GSTM1。S-III (n= 38)与增殖/转移相关,包括PKM、细胞间黏附分子-1、ACTN1、ITGB5和ANXA1。S-II具有S-I和S-III的中间特征。值得注意的是,S-I组患者的临床结局最好,而S-III组患者的预后最差,表现为最低的总生存率和最高的复发率(图4B-C)。与目前的临床知识一致,S-I亚组早期HCC患者较多,而S-III亚组患者分化程度较低,肿瘤侵袭性更强,肿瘤分期更晚,表现为较S-I和S-II亚组患者更高的AFP程度、更大的肿瘤大小、血管侵犯、TNM T分期、TNM分期、微血管侵犯和复发(图4A)。
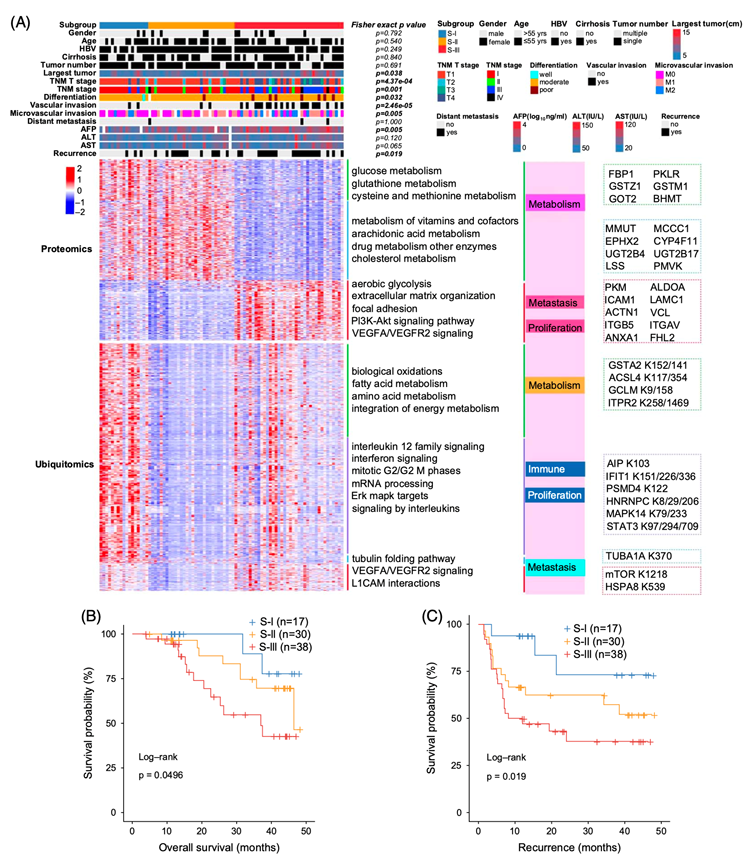
图4. HCC的分子分型及其与临床预后的关系。
(A) 热图显示有85个HCC样本聚集
分为3个亚型,S-I,S-II和S-III 以及其相关的临床特征。(B-C)生存分
05
S-III肿瘤中特征蛋白和通路的泛素化和HCC的潜在预后生物标志物
为了获得与S-III相关的细胞通路的特征,他们整合了所有85例HCC患者的多组学数据。差异表达分析和通路富集分析显示,S-III亚组中促癌蛋白(如PKM、细胞间黏附分子-1、MMP9)表达上调,促癌通路(如葡萄糖代谢和转移相关VEGFA/VEGFR2信号通路、上皮-间充质转化通路)激活,抑癌蛋白(如DHRS3、ADH1B、MAOA、ALDH2)表达下调。肝癌侵袭性S-III亚群(如GAPDH, HSPA8, CTNNB1和mTOR)的泛素化蛋白组学特征在分子水平上的聚合进一步验证了S-III肿瘤的侵袭性特征。相比之下,侵袭性较低的亚组(包括DHRS3、ADH1B和ALDH2)的蛋白质组学特征在S-I和S-II亚组的HCC中富集。S-III肿瘤富集蛋白显著富集于代谢相关、增殖相关或转移相关通路,包括糖酵解、谷氨酰胺分解、Wnt/β-catenin、蛋白酶体、细胞周期、mTOR和上皮-间充质转化信号通路(图5A)。
在S-III肿瘤中显著表达的前31种蛋白及其对应的56种泛素化化修饰蛋白(ubiquitoproteins)(图5B)。他们利用癌症基因组图谱数据集和GSE14520 HCC队列分析了这些显著表达的蛋白在mRNA水平的基因表达。为了确定潜在的S-III预后生物标志物,他们分析了18对配对的HCC患者的蛋白和泛素化修饰蛋白的表达,以及85例HCC患者的生存和复发的相关性。在肿瘤和ANT中,4种蛋白(TUBA1A、BHMT2、BHMT和ACY1)及其泛素化修饰蛋白(TUBA1A K370、BHMT2 K331、BHMT K93和ACY1 K332)的表达发生了显著变化,并且与OS和DFS强相关(图5C-F)。此外,TUBA1A K370的表达在S-III中显著下调,而其他3种泛素化修饰蛋白的表达在S-III中上调(图5G)。相比之下,TUBA1A蛋白在S-III中上调,而其他3种蛋白在S-III中下调。此外,TUBA1A与不良预后呈正相关,而其他3种蛋白与临床结局呈正相关(图5H)。相反,K370位点泛素化TUBA1A与不良预后呈负相关,而其他3种泛素化蛋白与临床结局呈负相关(图5H)。这些结果表明TUBA1A,BHMT2,BHMT和ACY1具有较高的预后风险评分,提示靶向这些蛋白或其修饰形式可能有利于未来的临床治疗。
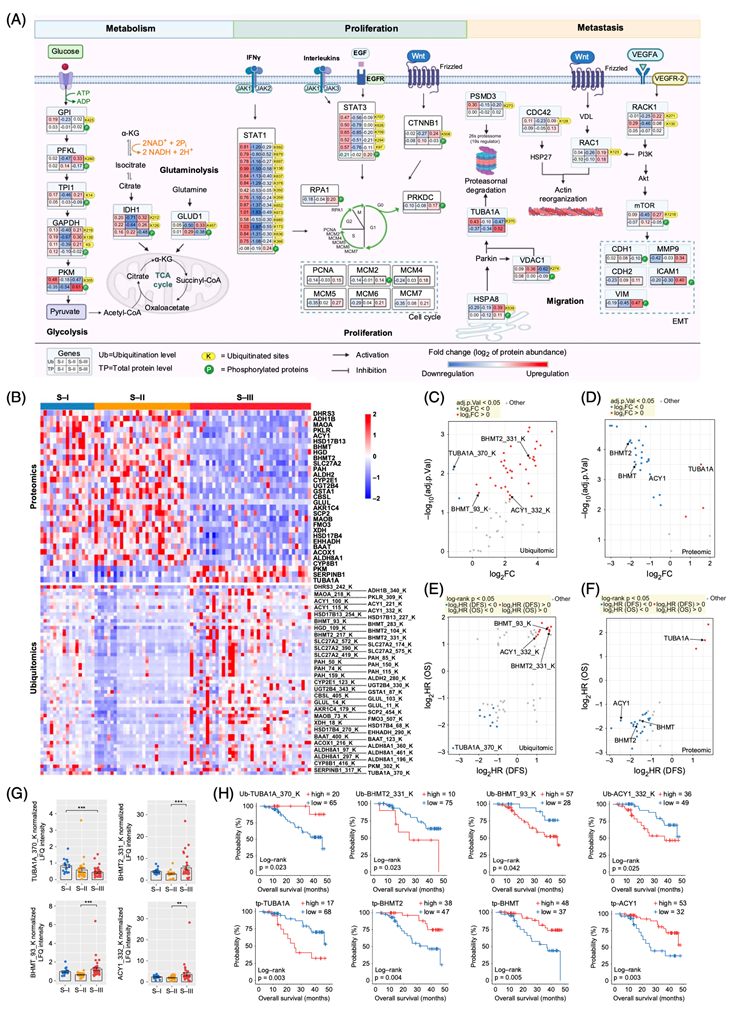
图5. S-III肿瘤的蛋白质组学和通路特征以及潜在预后生物标志物的鉴定和验证。
(A) 基于整合蛋白质组学分析的与S-III肿瘤相关的富集通路概述。(B) 通过蛋白质组学和泛素化蛋白组学分析,热图显示了31个总蛋白和56个泛素化修饰蛋白鉴定为S-III生物标志物。(C-D) 点图显示了不同蛋白表达。(E-F) 基于泛素化蛋白组学和蛋白质组学绘制56个泛素化修饰蛋白或31个总蛋白与OS或DFS的关系的点图。(G) 各亚组中泛素化修饰蛋白TUBA1A K370、BHMT2 K331、BHMT K93和ACY1 K332的表达。(H)生存分析。
06
去泛素化TUBA1A K370驱动肝癌增殖和转移
据报道,TUBA1A在胃癌中的高表达与更具有侵袭性的临床病理特征相关,但TUBA1A是否以及如何调控HCC尚不清楚。TUBA1A表达水平在HCC肿瘤的mRNA和蛋白水平均显著增加(图6A-B)。在人肝癌细胞中,TUBA1A高表达促进细胞增殖和迁移,而TUBA1A敲低则抑制这些作用。微管是由微管蛋白(包括TUBA1A)组成的细胞骨架微丝,在细胞分裂、增殖和迁移中发挥重要作用。TUBA1A过表达加速了微管组装和有丝分裂,促进了F-肌动蛋白聚合和细胞运动,从而导致肝癌细胞增殖和迁移。
目前尚不清楚TUBA1A的致癌活性是否与其在K370的泛素化有关。TUBA1A基因的K370位点在不同物种中进化保守。为了确定K370泛素化是否参与了TUBA1A的稳定性,他们将TUBA1A中的K370突变为精氨酸(K370R)。放线菌酮追踪实验显示,与野生型TUBA1A相比,突变型TUBA1A的K370R延长了Hep3B细胞中TUBA1A的半衰期,表明K370R突变可以抑制TUBA1A的蛋白酶体降解。正如预期的那样,TUBA1A K370R突变在体外和体内促进HCC细胞增殖和成瘤,并且这种作用比TUBA1A野生型强得多(图6C-F)。同样,与TUBA1A野生型相比,TUBA1A K370R突变显著促进了HCC的迁移和肺转移(图6G-H)。为了进一步评估TUBA1A在HCC中的潜在致癌作用,他们首先分析了总蛋白和泛素化TUBA1A水平与临床病理特征之间的关系。TUBA1A高表达及其在K370的去泛素化与血管侵袭和复发显著相关。这些结果表明,K370去泛素化在tuba1a介导的肝癌发生和肺转移中发挥了有效的促进作用。
泛素特异性蛋白酶(USP)家族是去泛素化酶的最大亚类,在底物去泛素化及其功能的调节中起着至关重要的作用。为了进一步验证USP家族的哪个成员参与了TUBA1A的K370去泛素化,他们通过蛋白质组学分析了18对配对的HCC样本中的USP家族蛋白水平。USP7、USP14、USP24和USP39在肿瘤中高表达,只有USP7和USP14与HCC患者的OS呈负相关。然后,他们进行了免疫共沉淀(Co-IP)实验,发现只有内源性USP14可与异位表达的TUBA1A发生共沉淀。在Huh7和Hep3B细胞中,内源性TUBA1A可与内源性USP14共沉淀(图6I),这些结果表明USP14可能是TUBA1A的关键去泛素化酶。接下来,他们试图确定USP14是否控制TUBA1A K370去泛素化。体内泛素化实验表明,USP14敲低增加了野生型TUBA1A的多泛素化,但对其K370R突变体没有影响。在HEK293T细胞中(图6J),表明USP14促进TUBA1A K370去泛素化。此外,USP14敲低缩短了TUBA1A的半衰期,但没有缩短其K370R突变体在Hep3B中的半衰期,表明USP14促进TUBA1A的稳定性。在功能上,随后证实USP14敲低显著阻断了TUBA1A介导的Huh7和Hep3B细胞增殖和迁移(图6K-L)。总的来说,这些发现表明,在K370上依赖USP14的TUBA1A去泛素化促进TUBA1A的稳定性,并诱导HCC的增殖和转移。
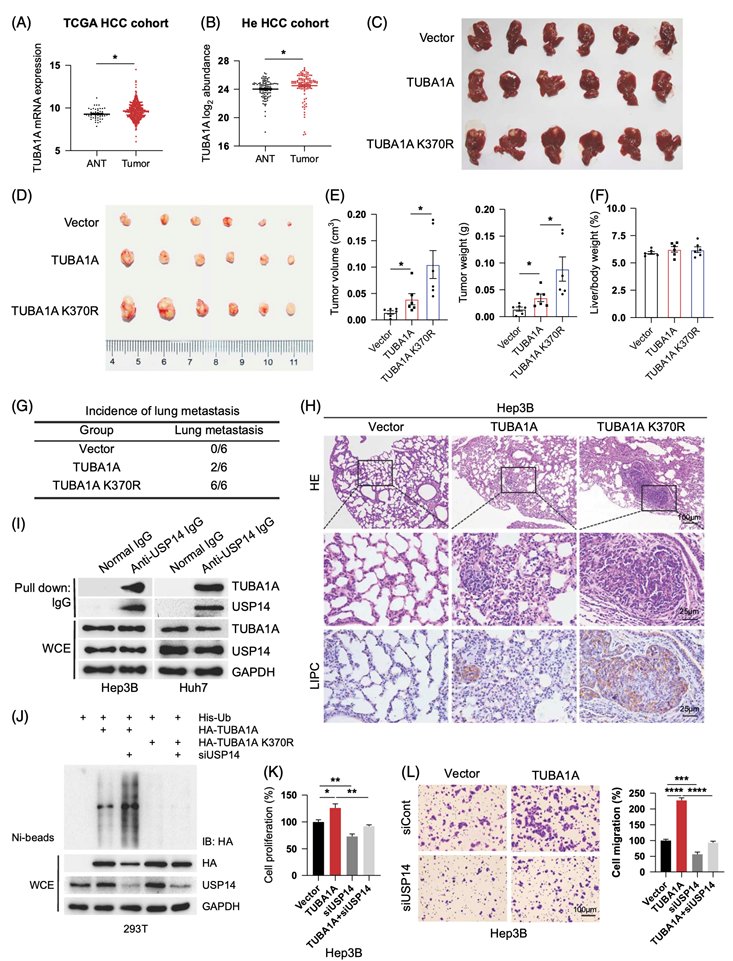
图6. TUBA1A K370去泛素化可促进HCC的增殖和转移。
(A-B)结合公开队列数据分析。(C-H)体内侵袭实验。(I) USP14与内源性TUBA1A的相互作用。(J) USP14敲低增加TUBA1A多泛素化。(K-L)细胞增殖实验和侵袭实验。
07
使用capivasertib和USP14抑制剂靶向TUBA1A可阻断HCC的肿瘤发生
USP14是蛋白激酶B (AKT)的底物,AKT促进USP14-S432磷酸化并增强其稳定性。目前,口服AKT小分子抑制剂capivasertib已批准用于治疗晚期乳腺癌患者,但capivasertib对USP14或TUBA1A高表达的HCC患者的疗效仍未知。因此,他们在HCC细胞系和高表达TUBA1A的小鼠肿瘤中评估了通过AKT抑制剂(capivasertib)和USP14抑制剂(IU1-248)靶向TUBA1A(图7A)。IU1-248显著抑制了USP14在S432的磷酸化,降低了TUBA1A野生型的表达,但未降低其K370突变型的表达,并且AKT抑制剂capivasertib抑制了AKT活化,并进一步增强了这些效应。在HCC- hep3b细胞系中,capivasertib联合IU1-248显著抑制HCC细胞增殖和迁移。在异种移植模型中,与TUBA1A K370R突变组相比,capivasertib联合IU1-248降低了TUBA1A野生型组的肿瘤体积和肿瘤重量(图7B-D)。各组小鼠的体重无显著差异(图7E)。为了进一步验证capivasertib和IU1-248对HCC增殖和迁移的抑制作用是否归因于促进TUBA1A多泛素化和降解,体内泛素化实验表明,capivasertib和IU1-248联合应用显著促进TUBA1A多泛素化,而K370R突变阻断了这一作用(图7F)。为了证实这些抑制剂促进TUBA1A降解,他们分析了capivasertib、IUI-248或两者联合治疗后TUBA1A的半衰期。放线菌酮追赶实验显示,与对照组相比,capivasertib联合IU1-248缩短了TUBA1A野生型的半衰期,而对其K370R突变型的半衰期无影响。总体而言,这些结果提示,抑制AKT和USP14可能通过诱导TUBA1A K370多泛素化和降解来治疗TUBA1A高表达的HCC患者,这为HCC患者提供了一种新的潜在治疗方法(图7G)。
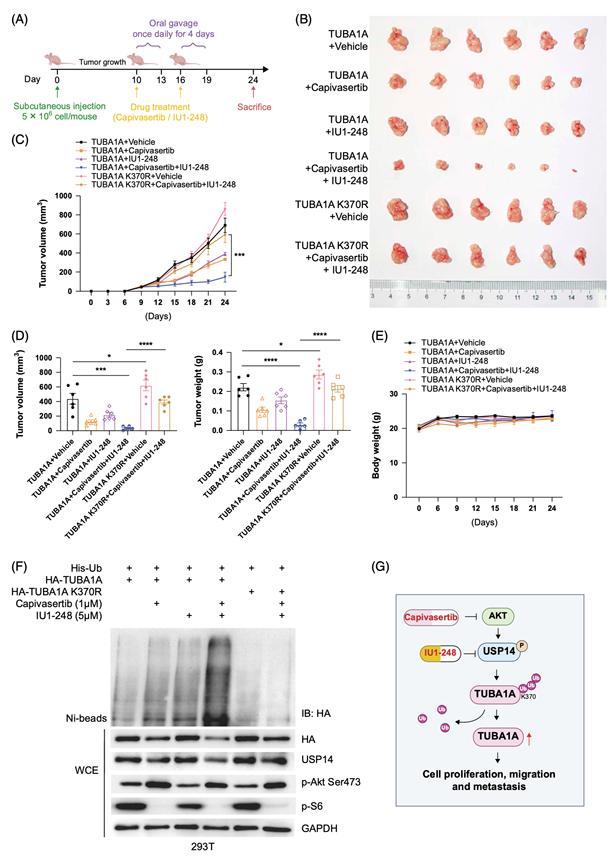
图7. 通过capivasertib和USP14抑制剂靶向TUBA1A阻断HCC的肿瘤发生。
(A)实验方案。(B-E) AKT抑制剂capivasertib联合USP14抑制剂IU1-248阻断TUBA1A介导的Hep3B-HCC小鼠异种移植瘤生长。(F)免疫印迹分析。(G) 机制示意图。
+ + + + + + + + + + +
结 论
本研究对 85 名 HCC 患者的肿瘤和邻近正常肝组织进行了全面的蛋白质组学、磷酸化蛋白质组学和泛素化蛋白组学分析。HCC 显示药物靶标 CBR1-S151 和 CPNE1-S55 的过度表达。COL4A1、LAMC1 和 LAMA4 在无病生存率低的患者中高表达。HCC 的磷酸化蛋白质组学和泛素化蛋白组学特征揭示了代谢和转移之间的相互影响。泛素化蛋白组学预测了不同的预后并阐明了 HCC 亚型特异性蛋白质组学特征。生物标志物 TUBA1A、BHMT2、BHMT 和 ACY1 的表达表现出不同的泛素化水平并显示出较高的预后风险评分,表明针对这些蛋白质或其修饰形式可能对未来的临床治疗有益。TUBA1A K370 去泛素化可导致严重的肝细胞癌,并可标记为一种侵袭性肝细胞癌亚型。TUBA1A K370 去泛素化至少部分归因于肝细胞癌中蛋白激酶 B 介导的 USP14 激活。值得注意的是,靶向 AKT-USP14-TUBA1A 复合物可促进 TUBA1A 降解并阻止体内肝肿瘤形成。
+ + + + +



