

 English
English文献解读|Science(44.7):基于肾脏多组学的基因评分卡揭示了编码变异和调控变异的趋同
✦ +
+
论文ID
原名:Kidney multiome-based genetic scorecard reveals convergent coding and regulatory variants
译名:基于肾脏多组学的基因评分卡揭示了编码变异和调控变异的趋同
期刊:Science
影响因子:44.7
发表时间:2025.02.07
DOI号:10.1126/science.adp4753
背 景
全球有超过 8 亿人患有肾脏疾病,每年有近 100 万人死于肾衰竭。肾脏功能具有高度遗传性,主要受常见遗传变异的影响。全基因组关联研究 (GWAS) 绘制了这些变异与疾病之间的关联,但 GWAS 识别出的变异中超过 90% 位于非编码基因组区域。这给确定其靶基因和调控功能带来了巨大挑战,这一难题称为“变异到基因”或“变异到功能”问题。
实验设计
结 果
01

来自 220 万个体的基因发现
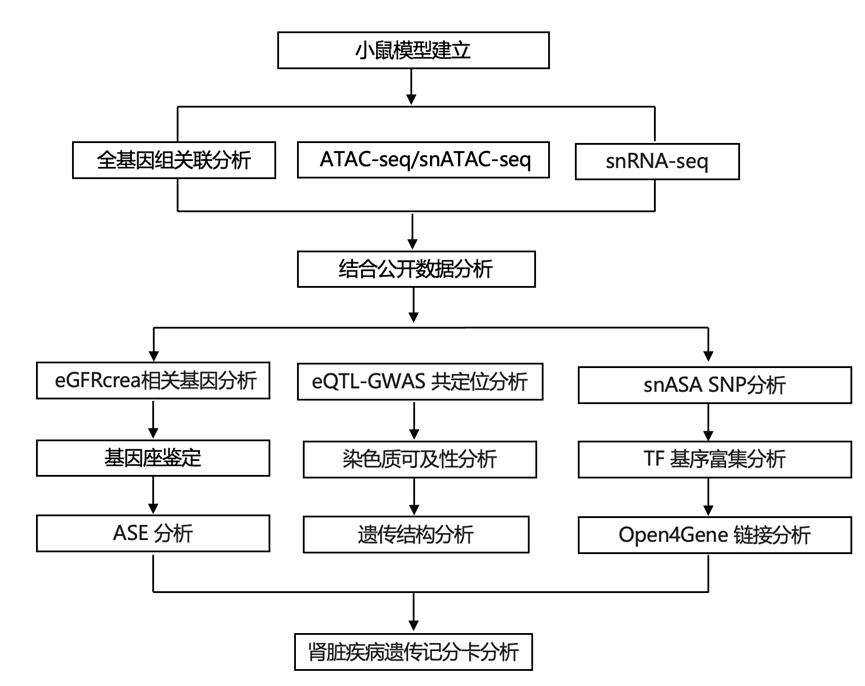
研究团队整合了来自 19 个队列的数百万个体的数据,对肾功能进行了遗传分析,以基于血清肌酐的估计肾小球滤过率 (eGFRcrea) 来衡量。首先,他们绘制了 9 个队列(MVP、TPMI、All of Us、MGB、GHS、BioMe、PMBB、eMERGE-III 和 CCPM)中 861840 名个体(包括 639074 名欧洲血统参与者、136620 名东亚血统参与者和 34879 名非洲血统人群)的遗传关联。然后,他们结合了来自 10 项先前发表的研究(CKDGen、Pan-UK Biobank、HUNT、BioVU、MGI、TOPMed、UKHLS、PAGE、SUMMIT 和 BBJ)的 1413622 名个体的汇总数据,进行了Meta分析。通过使用 METAL对z分数进行样本量加权meta分析,他们开发了一个全基因组 eGFRcrea 关联目录,涵盖欧洲血统、东亚血统和非洲血统人群以及所有血统(图 1A)。为了评估这些 GWAS meta分析中的人口分层程度,他们采用了连锁不平衡 (LD) 得分回归模型,LD 得分截距范围为 0.912 至 1.109,衰减率较低,这与多基因性下的预期一致,这表明没有明显的由于人口分层而导致的过度膨胀。
在涵盖 13700391 个变异的多祖先 GWAS 中,他们鉴定出 99595 个全基因组显著变异,排除了 12895 个来自主要组织相容性复合体基因座的显著变异。此外,他们还鉴定出 1026 个独立基因座,其中 97 个基因座距离任何报道的 eGFRcrea 相关变异位点至少 500 kb(图 1B)。这些 eGFRcrea 相关基因位点中的大多数(86.8%,891/1026)已通过至少一种与补充肾功能测量、血清胱抑素 C 为基础的 eGFR (eGFRcys)和血尿素氮(BUN)相关的变异体已证实为肾功能基因位点(图 1C)。这些独立的(包括新发现的)基因座在与肾脏发育和功能关键基因相关的基因组区域附近显著富集。97 个新基因座中超过一半(42/78)的优先基因先前已报道与肾脏功能或疾病有关,包括叉头框 C2 (FOXC2)、多囊蛋白 2 (PKD2)和盐诱导激酶3(SIK3)(图 1G)。已识别基因座的数量随样本量增加而增加,然而,在欧洲血统 GWAS 中,当样本量超过 150 万时,这种上升趋势开始减弱(图 1D)。
为了确定 eGFRcrea 基因座上的推定因果变异,他们使用 CARMA(一种用于meta分析精细定位的贝叶斯模型)对欧洲血统和多血统 GWAS 分别进行了精细定位。在欧洲血统 GWAS 中,他们划定了 1818 个集合,包含 99% 的可信区间,当纳入 PolyFun 注释时,其中 45.7%(830/1818)具有单个推定因果变异(图 1E)。这些可信集包括 8901 个推定的因果变异,其中非同义变异、肾脏增强子和启动子显著富集(图 1F)。这些集合中的大多数(82.0%)也在多祖先 GWAS 中观察到(图 1E)。例如,在 7 号染色体基因座 (q32.1) 处,他们鉴定出两个统计上不同的信号,它们具有可信集和不同的调控特征,一个位于 IRF5 (干扰素调节因子 5)基因启动子附近,与IRF5的数量性状基因座 (QTL)共定位(图 1G)和另一个与增强子相邻并与TNPO3(转运蛋白 3)基因的等位基因表达共定位。此外,他们与转录表达 QTL (eQTL)和DNA 甲基化 QTL (meQTL)数据集的共定位分析导致优先纳入 387 个基因和 4363 个 CpG 位点,它们表现出与肾功能共同的遗传倾向。
为了评估肾脏疾病与其他疾病之间的共同遗传基础,他们使用 HyPrColoc对肾小球滤过率 (eGFRcrea)与 23 个可能与肾功能相关的 GWAS 性状进行了全基因组多性状共定位分析。该分析鉴定了 351 个基因座的多效性图谱,其中 eGFRcrea 与其他性状共定位,区域概率 >0.8,其中 64.4% (226/351) 与已知的肾功能性状共定位。每个信号的共定位数量各不相同,从 4 个空腹血糖基因座到 183 个 eGFRcys 基因座不等(图 1H)。在这些基因座中,eGFRcrea 在 164 个基因座上与单一性状共定位,而 187 个基因座与多个性状共定位,包括GCKR基因座(15 个性状,包括 2 型糖尿病)、FGF5(11 个性状,包括收缩压)和APOE(5 个性状,包括父母长寿)。
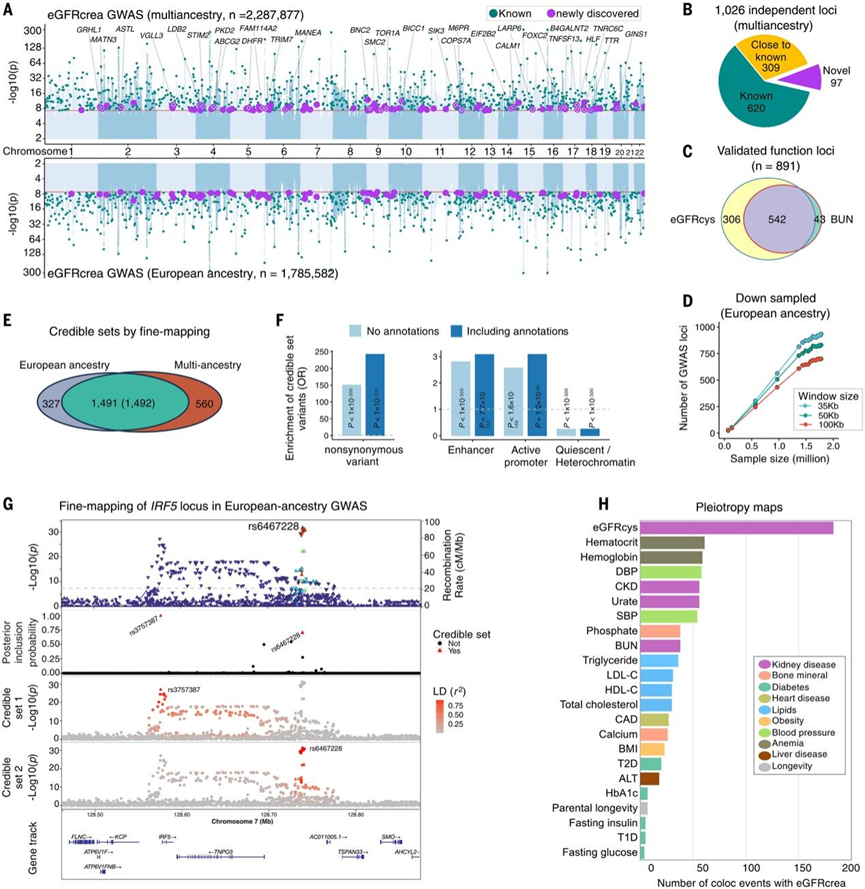
图1. 220 万个体的 eGFRcrea GWAS。
(A) 2287877 名多血统个体和 1785582 名欧洲血统个体的 eGFRcrea GWAS 的曼哈顿图。(B) 在多血统 GWAS 中确定的已知和新的独立基因座的比例。(C) 至少有一个经 eGFRcys 和 BUN 验证的变异的独立基因座的数量。(D) 在欧洲血统的下采样 GWAS 中识别的 GWAS 基因座数量。(E) 通过在 2287877 名多血统个体和 1785582 名欧洲血统个体的 eGFRcrea GWAS 中精细映射可信集的重叠。(F) 非同义变体、肾脏增强子和活性启动子中可信集变体的富集。(G) 通过在欧洲血统 GWAS 中的IRF5基因座中精细映射可信集的基因座。(H) eGFRcrea 和其他性状的多效性图。
02
人类肾脏中的等位基因特异性表达
为了阐明遗传因素对肾脏内基因表达的影响,他们对两个不同的肾脏区域进行了等位基因特异性表达 (ASE) 分析:肾小管(样本量为 613 人)和肾小球(样本量为 522 人)。该分析使用 RASQUAL进行,并根据技术和临床变量以及表达残差的概率估计 (PEER) 因子进行了调整(图 2A)。他们鉴定了 10398 个 ASE 基因,其中 8573 个位于肾小管,8725 个位于肾小球(图 2B)。作为验证,他们还在同一数据集中使用 MatrixeQTL 进行了典型的 eQTL 定位。两种方法估计的效应大小显著相关,RASQUAL 识别了 389 个典型 eQTL 定位未报告的额外基因(图 2C)。大约 60% 的 ASE 基因在 AlleleDB(1000 基因组变异目录的等位基因特异性注释)中表现出等位基因特异性表达,而仅通过典型 eQTL 识别的基因中这一比例为 45.6%(图 2C)。在这些 ASE 基因中,1773 个(17.1%)此前未在肾脏 eQTL 研究中发现。该子集包括已知在肾脏功能中发挥作用的基因,例如甘氨酸脒基转移酶(GATM)和 HNF1 同源框 A (HNF1A)(图 2D)。此外,他们观察到次要等位基因频率 (MAF) < 10% 的变异往往对基因表达表现出更大的影响,这与之前的 eQTL 研究一致。
此外,他们使用 ASE 关联对 eGFRcrea GWAS 中发现的变异进行优先排序,这导致提名了 32610 个变异(图 2E)。为了进一步完善潜在致病基因列表,他们进行了共定位分析,并突出显示了 531 个 ASE 关联与 eGFRcrea GWAS 信号共定位的基因(图 2F)。这些基因在代谢过程中显著富集。而大多数(76.7%,407/531)ASE-GWAS 共定位基因也表现出 eQTL-GWAS 共定位(图 2F),124 个基因在 eQTL 和 eGFRcrea GWAS 之间没有显示共定位(图 2F),例如,SLC22A2(又名OCT2,一种肾功能基因)表现出显著的ASE特征;然而,仅在ASE和eGFRcrea GWAS之间观察到共定位(图2G),强调了 ASE 分析在揭示与肾功能相关的信号方面的有效性。
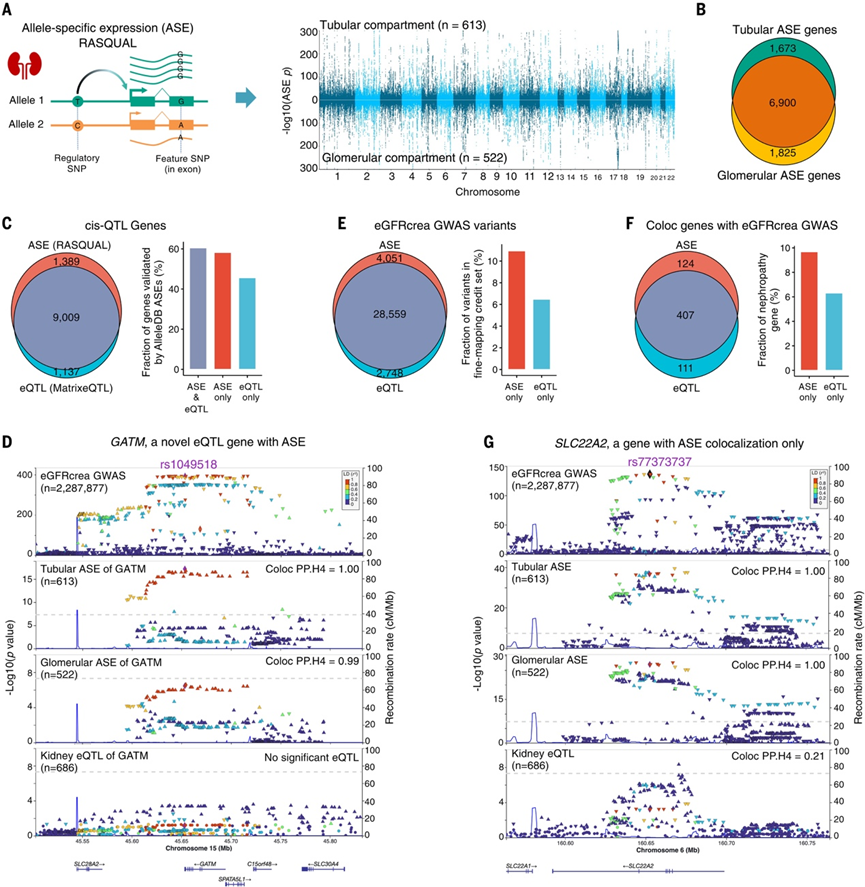
图2. 肾小管和肾小球区室的 ASE 分析。
(A) 肾小管和肾小球区室中 ASE 分析的分析方案和曼哈顿图。(B) 使用 RASQUAL 在肾小管和肾小球区室中鉴定的 ASE 基因的重叠。(C) 同一数据集中 RASQUAL 鉴定的 ASE 基因数量和 MatrixeQTL 鉴定的 eQTL 基因数量(左)以及 AlleleDB ASE 验证的基因分数(右)。(D) GATM基因座中 eGFRcrea GWAS、肾小管 ASE、肾小球 ASE 和肾脏 eQTL 的位点缩放。(E) 与 ASE 或 eQTL 相关的 eGFRcrea 相关变异的数量(左)以及精细映射可信 eGFRcrea GWAS 集中 ASE 或 eQTL 仅有的变异的比例(右)。(F) ASE 或 eQTL 与 eGFRcrea GWAS 共定位的基因数量(左)以及报告为肾病基因的 ASE 或 eQTL 仅有的基因的比例(右)。(G) SLC22A2基因座中 eGFRcrea GWAS、管状 ASE、肾小球 ASE 和肾脏 eQTL 的轨迹缩放。
03
大量肾脏中等位基因特异性染色质的可及性
染色质可及性对于基因调控至关重要,但基因型对肾脏内染色质可及性的影响尚未确定。为了填补这一空白,他们对人类肾脏样本进行了全基因组测序和高通量转座酶可及染色质测序 (ATAC-seq)。最初,他们建立了染色质可及性的综合图谱,其中包括 446385 个开放染色质峰,在 82 个大量肾脏样本[基于ATAC的等位基因特异性可及性的批量测定 (bASA) ]中绘制了等位基因特异性染色质可及性图谱。这些分析确定了 19083 个峰值,其中 523700 个 SNP 的次等位基因频率(MAF) > 5%,这些峰值与 RASQUAL 的染色质可及性变化有关。
与非 bASA 峰相比,人类肾脏 bASA 峰在鉴定为肾脏增强子的区域中显著富集,比值比 (OR) 为 1.7,在活性启动子区域中显著富集,比值比 (OR) 为 2.2(图 3B)。上面鉴定出的大多数 bASA SNP 位于相关峰的 50 kb 范围内。他们发现 8317 个 bASA 峰与局部(峰内)和远端(峰外)SNP 均有关联,它们对同一峰产生一致的影响。MAF < 10% 的变异对染色质可及性的影响更大,与本研究中等位基因特异表达 (ASE) 分析的结果一致。此外,bASA 的效应大小与甲基化 QTL 呈负相关,与 ASE 呈正相关,表明开放染色质对基因表达具有调控作用(图 3C-D)。此外,他们使用 bASA 关联对受 eGFRcrea GWAS 变异影响的开放染色质峰进行优先排序,在 445 个 eGFR GWAS 基因座的 997 个 bASA 峰中发现了 20035 个与 eGFRcrea 和人类肾脏染色质可及性相关的单核苷酸多态性(SNP)(图 3E),大多数 bASA SNP(88.9%) 也与 DNA 甲基化和等位基因特异性表达有显著关联。
为了研究开放染色质和肾功能之间的共同遗传结构,他们进行了共定位分析,确定了 209 个位点,其中 eGFRcrea GWAS 信号与 268 个峰的可及性共定位(图 3E)。值得注意的是,其中大多数(82.3%)也与 meQTL 和/或 ASE 共定位(图 3E)。具体而言,在 104 个基因的 79 个位点上,eGFRcrea 与所有三种分子性状(bASA、meQTL 和 ASE)共定位,其中包括先前与肾脏疾病相关的基因(例如UMOD和UNCX)以及潜在的新疾病基因(例如USP24和ACSM2A)。例如,USP24基因座处的遗传变异与共有变异 rs2802865 处的 eGFRcrea、开放染色质、CpG 甲基化和基因表达显著相关(图 3F)。 大多数 (91.9%, 192/209) eGFRcrea~bASA 共定位位点已通过补充肾功能测量(eGFRcys 和 BUN)验证为肾功能位点,强调了遗传变异和表观遗传调控元件在影响肾功能方面的相互交织。
图3. 人类肾脏中的 ASA 分析。
(A) 人类肾脏中 bASA 分析的分析工作流程和曼哈顿图。 (B) 使用组蛋白修饰估计的肾脏染色质状态中 bASA 峰的富集。(C) 肾脏 bASA(x 轴)和 meQTL(y 轴)之间效应大小的关联。(D) 肾脏 bASA(x轴)和管状 ASE(y轴)之间效应大小的关联。(E) 使用bASA结合 ASE 和 meQTL 对 GWAS 变体和基因座进行优先级排序。(F) USP24基因座中 eGFRcrea GWAS、肾脏 bASA、meQTL 和小管 ASE 的调控元件和位点图谱。
04
肾脏细胞类型中等位基因特异性染色质的可及性
肾脏有 30 多种细胞类型,为了辨别基因型对特定肾脏细胞类型染色质可及性的影响,他们使用单细胞核 ATAC 测序 (snATAC-seq) 技术对 30 例人类肾脏中的单个细胞进行了分析,汇编了一份涵盖 237274 个细胞的开放染色质图谱,涵盖 19 种细胞类型,每种类型的中位数为 5302 个细胞。在近端小管特异性开放染色质峰内,eGFRcrea GWAS 变体显著富集(图4A)。然后,他们通过进行基于 snATAC-seq 的ASA (snASA) 分析,评估了等位基因对每种细胞类型内染色质可及性的影响(图 4A),总共鉴定出 25766 个 SNP,它们在至少一种细胞类型中表现出显著的 snASA(图 4B)。大多数 snASA SNP位于开放染色质峰内(图 4C),并且非峰值 snASA SNP 显示出明显高于非峰值区域的改组 SNP 的读取覆盖率。此外,他们观察到 MAF < 10% 的变异往往对染色质可及性表现出更大的影响,这与 ASE 和 bASA 分析一致。
大多数 snASA SNP特定于单个细胞类型,而 3615 个 snASA SNP 在至少两种细胞类型中是共有的(图 4D)。然后,通过分析前面提到的 82 个肾脏样本中 snASA 和 bASA 中均测试过的 18727 个常见 SNP,评估了 snASA 识别新调控变异的潜力。虽然大多数(68.4%)跨细胞类型常见的 snASA SNP 也能通过 bASA 检测到,但很大一部分(58.7%)细胞类型特异性 snASA SNP 未显示显著的 bASA 关联(图 4E),表明 snASA 在识别细胞类型特异性调控变异方面具有优势。此外,他们观察到等位基因效应大小在不同细胞类型之间以及 snASA 和 bASA 之间呈强正相关性(图 4F),这些相关性表明调节染色质可及性的强大生物学机制。例如,位于SLC45A4基因内含子中的变体 rs10875467 的等位基因 C 与大量肾脏样本和各种细胞类型(包括近端小管和亨利氏环)中染色质可及性降低显著相关。
为了研究在肾脏细胞类型中介导等位基因特异性染色质可及性的转录因子 (TF),他们对每种细胞类型特有的 snASA SNP 进行了转录因子(TF)基序富集分析,鉴定出 29 个 TF 基序优先由 snASA SNP 破坏,富集分数 >3 (图 4G)。在这些基序中,一些表现出细胞类型特异性破坏,例如近端小管中的 HNF4A 和 HNF4G 基序以及亨利氏环中的 ESRRB 和 ESRRA 基序。其他基序,例如 FOSL2 和 NFKB1 基序,在受损的近端小管细胞中富集。此外,他们发现了在不同细胞类型中具有共同富集的基序,例如与 CCCTC 结合因子 (CTCF) 结合的基序,CTCF 是一种参与调节三维基因组结构的结构蛋白(图 4G)。
在确定并描述了不同肾细胞类型中染色质可及性的等位基因不平衡后,他们接下来评估了 snASA SNP 是否能介导基因型对 eGFRcrea 的影响。在 337 个 GWAS 基因座中,669 个 snASA SNP 与 eGFRcrea 显著相关,其中包括DAB2、SHROOM3和SLC47A1基因座中与 eGFRcrea 关联的高概率变异(图 4D)。大多数(79.5%,532/669)snASA-GWAS SNP 已通过补充肾功能测量得到验证。这些 snASA-GWAS SNP 中相当一部分(63.4%,448/738)位于转录因子结合位点,频率明显高于与 snASA 无关的 GWAS SNP 中观察到的 18.8%(图 4H-I)。TF 富集分析进一步强调了CTCF、HNF4A、PAX5和KLF5在肾脏疾病发病机制中的潜在参与。这些研究结果表明,人类肾脏的等位基因特异性染色质可及性图谱有助于识别与肾脏功能有关的细胞类型和调控变异。
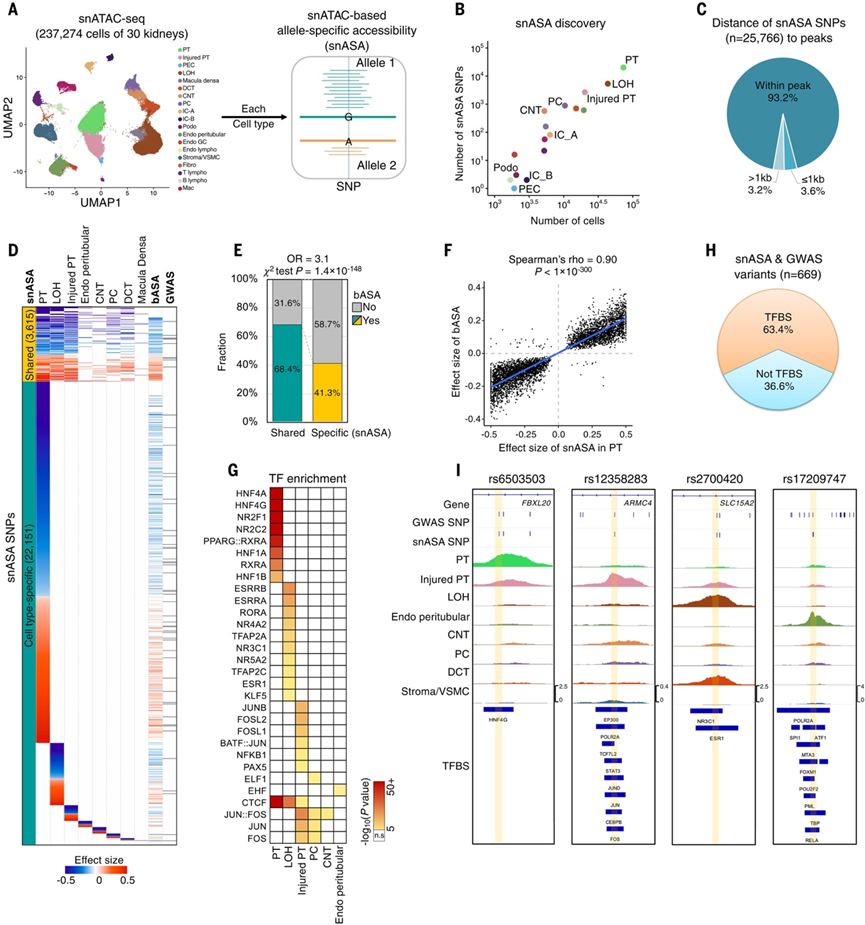
图4. 人肾脏单细胞水平ASA分析。
(A) 人类肾脏细胞类型中 snASA 分析的分析工作流程。(B) 每种细胞类型中鉴定出的细胞数(x轴)和 snASA SNP(y轴)。(C) 开放染色质峰内 snASA SNP 的比例。(D) 肾脏细胞类型(列)中细胞类型特异性和共有的 snASA SNP(行)。(E) 经 bASA 验证的细胞类型特异性和共享的 snASA SNP的比例。(F) PT snASA(x轴)和肾脏 bASAs(y轴)之间效应大小的关联。(G) 细胞类型特异性 snASA SNP 在 TFBS 上的富集。(H) snASA 和 GWAS 变体与 TF 结合位点重叠的比例。(I) snASA 和 GWAS 变体与细胞类型特异性的开放染色质峰和 TFBS 重叠的示例。
05
通过单细胞核多组学研究基因调控回路
接下来,他们使用单细胞核多组测序[单细胞转录组测序(snRNA-seq)和 snATAC-seq]同时分析了 9 个人类肾脏样本中的染色质可及性和基因表达。他们编制了一份全面的图谱,涵盖 19 种肾细胞类型的 62278 个细胞的染色质可及性和基因表达(图 5A)。为了精确定位参与基因激活的开放染色质区域,他们设计了一种名为 Open4Gene 的统计方法。该方法使用障碍负二项回归模型来分析染色质可及性与基因表达之间的关联(图 5A)。为了进行验证,他们将 Open4Gene 与两种广泛使用的单细胞多模态峰基因连锁方法 ArchR和 Signac一起应用,以分析基于 62278 个肾细胞的联合谱的一组 903472 个峰基因配对(涵盖 16239 个基因和 347480 个开放染色质峰,在 100 kb 顺式调控范围内)。与其他方法鉴定出的(排序靠前的)峰基因对相比,Open4Gene 鉴定出的这些(排序靠前的)峰基因对表现出显著更高的峰可及性和基因表达之间的相关得分。此外,模拟分析表明 Open4Gene 识别的假阳性率很低。这些发现表明 Open4Gene 是识别单细胞多模态数据集内功能性顺式调控元件的有效工具。
使用 Open4Gene 工具,他们为每种人类肾脏细胞类型描绘了一组明确的峰-基因关联。此过程产生了 35388 个不同的 Open4Gene 链接,其中 40.6% 与分析所有细胞类型的组合时获得的链接不同。在每种细胞类型中检测到的 Open4Gene 链接数量与该特定类型记录的细胞数量成正比(图 5B)。通过将每种细胞类型的发现与集体细胞分析的结果整合在一起,总共确定了 142452 个峰-基因链接,涵盖 125699 个峰,其中近 90% 与单个基因特异性相关(图 5C)。得出的 Open4Gene 关联表明每个基因有中位数为 8 个关联峰,其中与肾脏疾病有关的基因PKHD1具有最多的峰。此外,97% 的 Open4Gene 关联与其靶基因的表达呈正相关。Open4Gene β 值在不同细胞类型之间表现出高度相关性,强化了开放染色质在基因激活中的关键作用。
Open4Gene 通过识别位于 Open4Gene 峰内的 7137 个遗传变异的 1351 个靶基因,有助于将变异与基因表达和功能联系起来(图 5D)。位于这些 Open4Gene 峰中的遗传变异与肾功能表现出很强的相关性(图 5E)。他们观察到这些变异中有 80.2% (5723/7137) 是单个基因所独有的。有 253 个基因 10 个由更多 GWAS 变异靶向,这些基因在与肾脏系统生理异常相关的通路中表现出显著富集。这组基因包括之前与肾脏疾病相关的基因,例如LRP2、UMOD、DAB2、SLC34A1、PAX8和KLF5(图 5F-G)。对于文献中未知与肾脏关联的基因,他们重点研究了ACSM2A,它编码一种参与脂肪酸代谢的线粒体酶,并探索其在肾脏中的潜在功能。ACSM2A 的表达主要在近端小管中发现,并且似乎与开放染色质相关,如 Open4Gene 链接所示。利用来自 433 个人类肾脏的 RNA-seq数据,他们确定ACSM2A表达与肾功能显著正相关并与肾纤维化显著负相关。此外,ACSM2A表达在慢性肾病中显著降低,这一结果在三种小鼠模型中得到证实:单侧输尿管阻塞、叶酸肾病和顺铂模型。这些结果强调了 Open4Gene 框架在阐明疾病相关基因的调控机制和解决 GWAS 中的变异到功能问题方面的有效性。
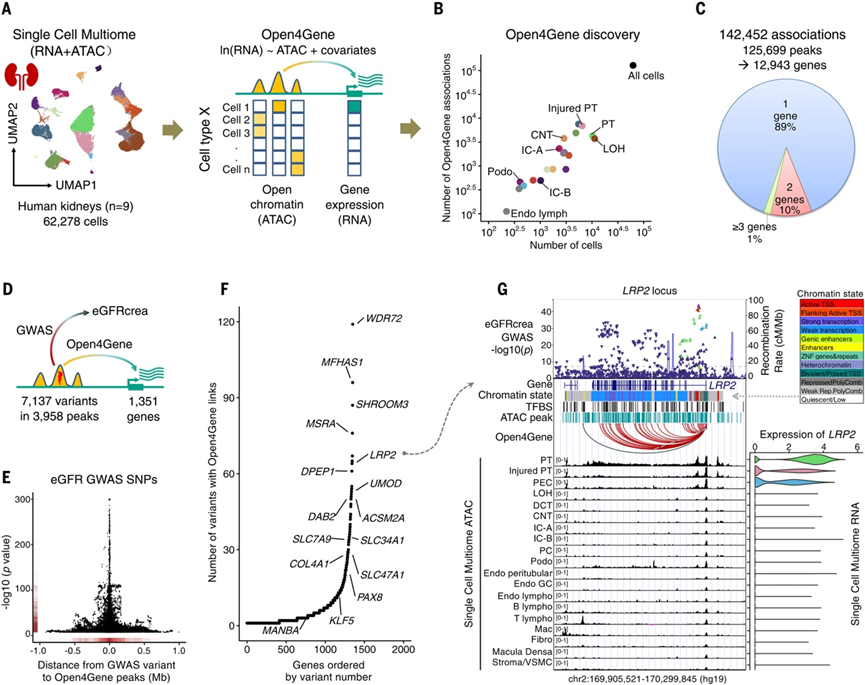
图5. 人类肾脏单细胞多组学 Open4Gene 关联将 GWAS 变体与目标基因联系起来。
(A) 人类肾细胞中单细胞核多组学 (RNA + ATAC) 和 Open4Gene 分析的分析工作流程。(B) 每种细胞类型中鉴定出的细胞数量(x轴)和 Open4Gene 链接(y轴)。(C) 针对不同基因和多个基因的开放染色质峰的分数。(D) 使用 Open4Gene 链接对 GWAS 变体的目标基因进行优先级排序。(E) eGFRcrea GWAS 强度(y轴)和 GWAS 变体到 Open4Gene 峰的距离(x轴)。(F) 使用 Open4Gene 链接为每个基因鉴定出的 GWAS 变体数量。(G) eGFRcrea GWAS、Open4Gene 链接和LRP2 基因座中的调控元件的轨迹缩放。
06
综合肾脏疾病基因记分卡概念,用于结合变异和基因优先排序
在本研究中,他们生成了大量用于注释调控变异的多组学数据集,包括体细胞和单细胞水平的等位基因特异性表达和染色质可及性;Open4Gene 链接,补充现有的 eQTL 数据集;以及 meQTL,全外显子组关联分析;此外,还使用了互补性状 GWAS 研究(eGFRcys 和 BUN)。在对不同基因和变异进行优先排序方面存在重要差异,没有单一工具或数据集能够实现从变异到功能的注释。因此,他们推断出一个“肾脏疾病遗传记分卡”,它包含所有可用的数据集和工具,并使用简单的加法方法来提名肾脏疾病基因和变异。他们纳入了八个数据集,包括 bASA、snASA 和精细映射可信集,以评估调控变异。此外,他们使用 12 个注释来评估基因的疾病风险,例如编码序列 (CDS)、全外显子组关联研究 (ExWAS)和全表型关联研究 (PheWAS),通过 12 种顺式关联分析,包括 Open4Gene 链接、ASE 和共定位分析,研究了非编码变异对靶基因的影响(图 6A)。优先变异的数量随着优先分数的增加而减少;然而,针对不同基因的变异比例明显增加,表明本研究的优先策略在精确定位真实的调控变异方面的有效性(图 6B)。
编码和调控变异都会增加患病风险,但它们在 GWAS 基因座中的集体存在仍未得到充分探索。为了填补这一空白,他们整合了多组学数据集,以优先考虑与肾功能和疾病相关的遗传变异(图 6A)。根据 eGFRcrea GWAS 变体与基因区域的接近程度对其进行了注释,发现其在蛋白质 CDS、5′ 非翻译区 (UTR)、3′ UTR 和内含子中存在显著富集(图 6C)。当他们根据染色质状态(由人类肾脏中的组蛋白修饰定义)注释 GWAS 变体时,注意到这些变体在基因增强子、活性启动子、增强子和其他调控元件中显著富集。然后,他们将重点缩小到编码变异。全面的GWAS 映射有助于识别与肾功能相关的 1363 种编码变异。GWAS 和 ExWAS 同时发现的常见编码变异与肾功能表现出高度相关的效应大小。通过使用变异效应预测器,他们精确定位了 83 种功能丧失变异(例如导致终止密码子增加、剪接区变异和移码突变的变异)、776 种错义变异和 505 种同义变异(图 6D)。在 782 个包含这些编码变异的基因中,超过 95% 尚未由 ExWAS 或 PheWAS 鉴定,后者主要关注 MAF < 10% 的罕见变异。在这些基因中,GWAS、ExWAS 和 PheWAS 共同鉴定出 21 个肾脏疾病相关基因,包括溶质载体基因(如SLC47A1和SLC6A19)、多配体受体(LRP2和CUBN)以及与多囊肾病有关的基因(PKD1、PKD2和PKHD1)。其中 289 个基因(占 782 个基因的 37.0%)具有多个编码变异,其中ALMS1(着丝粒和基底体相关蛋白)的编码变异数量最多(16 个),该基因的功能障碍可导致肾脏过滤功能逐渐下降。正如预期的那样,分析显示,虽然最常见的编码变异表现出较小的效应大小,但罕见的编码变异往往具有较大的效应大小。
他们集中研究了 316 个 GWAS 基因座中编码和调控变异确定的 601 个基因(图 6E)。这些基因中的大多数(93.8%,563/600)至少有一个变异体通过补充肾功能测量得到验证(图 6F)。601 个基因的功能富集分析表明它们在近端小管转运和肾脏特异性表型(如肾脏系统生理异常)中的作用。值得注意的是,其中 74 个基因是已记录的肾病基因,包括 10 个基因(PKD1、PKD2、PKHD1、CUBN、GATM、LRP2、SLC34A1、SLC34A3、SLC6A19和SLC7A9),这些基因已通过 ExWAS 和 PheWAS 验证。161 个基因的编码和调控变体趋于一致,其中包括 29 个已发表的导致敲除小鼠肾脏异常的基因(图 6F-G)。与非趋同编码变体相比,趋同的编码变体在 eGFRcrea GWAS 中表现出明显较低的次要等位基因频率,较高的效应大小和较高的显著性。
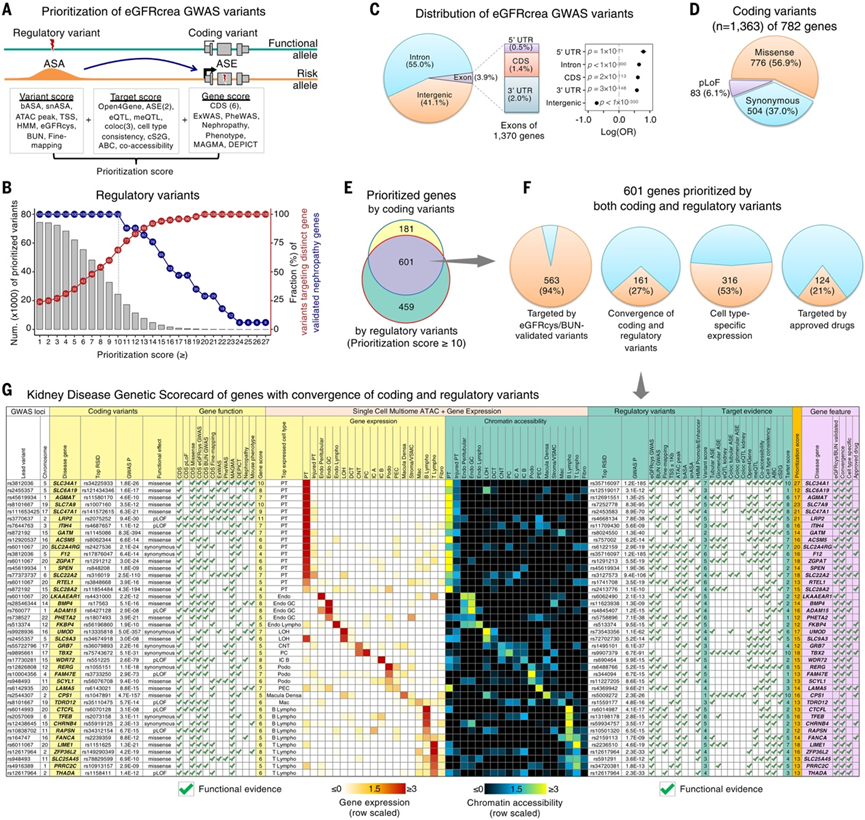
图6. 肾脏疾病遗传记分卡表明 GWAS 基因座中编码和调控变异的趋同。
(A) 通过整合肾脏多组学对 eGFRcrea GWAS 变异进行优先级排序的分析工作流程。(B) 通过整合肾脏多组学对调节变异进行优先级排序。(C) 位于不同基因组特征中的 eGFRcrea GWAS 变异数量(左)和x轴上使用对数转换 OR 的富集检验并标记每个特征的显著性(右)。(D) 注释为预测的功能丧失 (pLoF)、错义和同义词的编码 GWAS 变异的比例。(E) 按调节变异(优先级评分≥10)和编码变异优先排序的 eGFRcrea GWAS 基因座数量。(F) 经 eGFRcys 或 BUN 验证的基因数量,具有编码和调节变异的趋同性、具有细胞类型特异性表达、并由 FDA 批准药物靶向。(G) 编码和调节变异趋同的基因的肾脏疾病遗传记分卡(优先级评分≥12)。
+ + + + + + + + + + +
结 论
研究团队开展了一项多祖先 GWAS 研究,研究对象为 220 万名个体,研究以基于eGFRcrea 为衡量标准,研究对象为肾功能,确定了 1026 个(97 个之前未知)独立位点。通过绘制欧洲、东亚和非洲血统人群中与肾功能相关的常见 DNA 变异图谱,观察到欧洲人群中新发现的信号有所减弱,并强调了人群多样性对进一步发现的价值。本研究还分析了基因型对 700 多个肾脏和 237,000 多个细胞中等位基因特异性基因表达和调控贿赂的影响,开发了一种名为 Open4Gene 的统计方法,该方法确定了位于开放染色质区域内的 1351 个遗传变异靶基因。本研究引入了“肾脏疾病基因评分卡”概念,该概念整合了 32 种类型的数据来支持遗传信息并提名肾脏疾病的致病基因变异和基因。肾脏疾病基因评分卡优先考虑了 24437 个针对 1060 个基因的调节变异,还观察到特定基因中编码和调节变异的趋同,确定了 1363 个破坏 782 个基因的编码变异,其中 601 个基因也由调节变异靶向。值得注意的是,124 个基因已确定为适合 FDA 批准药物的靶向,为药物再利用和治疗开发提供了机会。
+ + + + +



