

 English
English文献解读|Psychiatry Res(11.30):综合多组学分析揭示重度抑郁症的肠道微生物群失调和系统紊乱
✦ +
+
论文ID
原名:Integrated multi-omics analysis reveals gut microbiota dysbiosis and systemic disturbance in major depressive disorder
译名:综合多组学分析揭示重度抑郁症的肠道微生物群失调和系统紊乱
期刊:Psychiatry Research
影响因子:11.30
发表时间:2024.02.27
DOI号:10.1016/j.psychres.2024.115804
背 景
重度抑郁症(MDD)涉及外周血液和肠道微生物群的系统性变化,但目前对MDD的发病机制缺乏完整的了解。
实验设计

结 果
01

患者特征和研究设计
本项研究包括一个由 99 名诊断患有 MDD 的个体和 50 名年龄匹配的健康对照(HC)组成的队列。MDD组的平均年龄为27.3岁,平均发病年龄为26.7岁。在这 99 名患者中,有 72 人经历了首次 MDD 发作。蒙哥马利-阿斯伯格抑郁评定量表(MADRS)、汉密尔顿抑郁评定量表-17 (HAMD-17)、抑郁症状快速量表(QIDS-SR16)和汉密尔顿抑郁评定量表-14 (HAMA-14)的平均得分分别为32.6、25.6、20.3和24.1。研究者团队通过16S rRNA测序和宏基因组学测量粪便样本的肠道微生物群,并分别通过靶向代谢组学和质谱流式细胞仪测量血液代谢物和免疫细胞表型,然后将获得的多组学数据进行生物信息学分析(图1A)。
为了研究与 MDD 相关的肠道微生物群的结构变化,他们首先利用 16S rRNA 测序。与HC组相比,MDD组在包括Chao1多样性在内的α多样性上没有明显变化(图1B)。然而,在beta多样性分析中,他们通过主坐标分析 (PCoA) 观察到MDD组和HC组之间存在显著差异(图1C)。对 MDD 患者和 HC 个体肠道微生物群组成的进一步检测揭示了不同的模式。在门水平上,MDD患者表现出较高丰度的拟杆菌门和较低丰度的厚壁菌门。同样,在属水平上,MDD 患者表现出较高的拟杆菌丰度和较低的粪杆菌属和巨单胞菌属丰度(图 1D)。为了比较MDD和HC之间肠道菌群组成的差异,他们采用了LEfSe分析。结果显示,不同的微生物类群与每个组相关。在MDD组中,Bacteroidaceae、Desulfovibrionaceae和Enterococcaceae的水平较高,而HC组中厚壁菌门 (Firmicutes)、Erysipelatoclostridiaceae、单胞杆菌科(Monoglobaceae)等的水平较高(图1E)。在属水平上,通过差异分析发现MDD和HC之间存在显著差异。MDD患者的肠球菌、Eubacterium_hallii_group、Oxalobacter和Intestinimonas水平较高,而Clostridium_sensu_stricto_1、Coriobacteriales_Incertae_Sedis和Family_XIII_AD3011_group的水平较低(图1F)。
为了确定 MDD 和 HC 之间的功能差异,他们利用了 MetaCyc 数据库,发现患有MDD的人表现出更高水平的乙醛酸循环和脂肪酸降解途径。相反,与HC相比,它们表现出较低水平的各种代谢途径,例如水苏糖降解、嘌呤核糖核苷降解和D-半乳糖降解I(图1G)。此外,他们研究了MDD中肠道微生物群功能与抑郁严重程度之间的关系,揭示了密切的相关性(图 1 H)。值得注意的是,adp-l-甘油-β-d-甘露-庚糖的生物合成与QIDS-SR16评分呈正相关。相反,一些肠道微生物群功能和途径,如gdp -甘露糖衍生的o-抗原构建块生物合成途径、黄素生物合成I、同化硫酸盐还原IV、5-氨基咪唑核糖核苷酸生物合成I和甲醛同化II与MADRS评分呈负相关。此外,黄素生物合成I也与HAMD-17评分呈负相关。这些发现表明,肠道微生物群功能的变化可能影响MDD的进展。
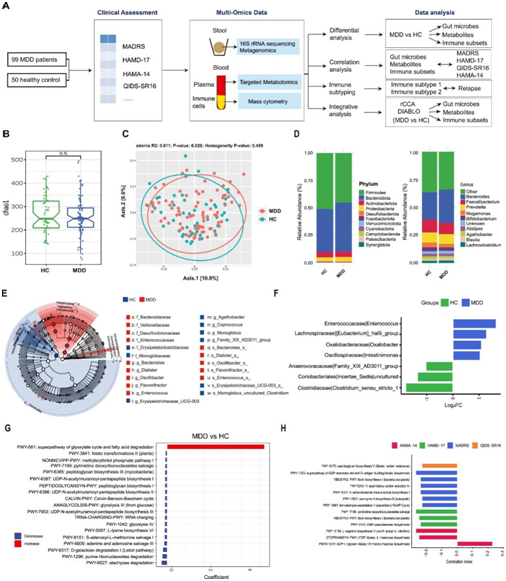
图1. MDD 肠道微生物群的研究设计和变化。
(a) 研究设计。临床诊断后收集MDD组和HC组的粪便和血液样本,通过多组学技术进行分析,并进行生物信息学分析。(B) HC 和 MDD 之间 Chao1 多样性指数的箱线图。(C) HC 和 MDD 的 PCoA 图。(D) HC 和 MDD 中每个分类水平(门、属)的富集类群的堆积条形图。(E) LEfSe 发现的显著差异富集的类群的分支图。(F) DESeq2 发现的 HC 和 MDD 之间属水平上显著差异富集的类群的条形图。(G) 通过分析宏基因组数据集,绘制HC和MDD之间显著差异富集的MetaCyc通路的条形图。(H) 通过分析宏基因组数据集,显示肠道微生物群功能(MetaCyc 通路)与 MDD 抑郁评分之间的相关性。
02
MDD患者血浆代谢物的变化及其与肠道微生物的相关性
他们采用有针对性的定量代谢组学方法分析了总共630种血浆代谢物。通过比较MDD和HC患者的血浆代谢物,他们观察到显著差异。在MDD中,一些小分子代谢物表现出明显的减少,包括脯氨酸-甜菜碱 (ProBetaine)、胆汁酸(GCA、GCDCA)、亚精胺、血清素、腐胺、胆碱和各种氨基酸(图2A)。相比之下,大多数脂质在MDD中增加,包括甘油三酯(tg) (22:6_34:1)、胆固醇酯(CEs)、神经酰胺(Cers)、己糖神经酰胺(HexCers)和鞘磷脂(SM),只有两种脂质,PC .ae.C38:1和lysoPC.a.C18:2,显示水平降低(图2B)。接下来,他们研究了血浆代谢物水平与抑郁症严重程度之间的关系,揭示了许多小分子代谢物与抑郁评分之间的显著相关性(图2C)。值得注意的是,精氨酸和脯氨酸代谢途径中的代谢物瓜氨酸(Cit)与QIDS-SR16、MADRS、HAMD-17和HAMA-14呈负相关。此外,该途径中的其他代谢物,如脯氨酸 (Pro)、精氨酸 (Arg) 和鸟氨酸 (Orn),也与抑郁症严重程度呈负相关,如Pro与QIDS-SR16、MADRS、HAMD-17呈负相关,而Arg、Orn与MADRS呈负相关。另一方面,某些代谢物显示出与抑郁得分呈正相关,这些包括对甲酚-so4、牛磺胆酸(TMCA)和硫酸乙醇胆酸(GLCAS) (QIDS-SR16),以及同型半胱氨酸(HCys)、FA(18:2)和FA(18:1) (MADRS)。此外,他们对血脂的分析也揭示了与抑郁评分的显著相关性。许多tg与QIDS-SR16和HAMD-17呈负相关,而磷脂酰胆碱(PC)与QIDS-SR16呈正相关。
为了探索血浆代谢物和肠道微生物群在MDD发展过程中的相互联系,他们进行了典型相关分析(CCA)来确定两者之间的相关性。他们的分析揭示了11种小分子代谢物与各种肠道微生物之间的显著关联(图2D)。例如,马尿酸(HipAcid)、GLCAS和对甲酚- so4分别与Coprococcus、[Eubacterium]eligens group和Agathobacter呈正相关。1-甲基组氨酸(1-Met-His)、α-氨基己二酸(α-aaa)、肌酐、犬尿氨酸、异亮氨酸、亮氨酸和3-甲基组氨酸(3-Met-His)与Lachnospiraceae_UCG-006、Granulicatella和Helicobacter等呈正相关,脂质和肠道微生物之间的联系相对有限(图3E)。
这些发现表明血浆代谢物的改变可能受到特定肠道微生物群的影响,突出了它们在MDD中的潜在相互作用。
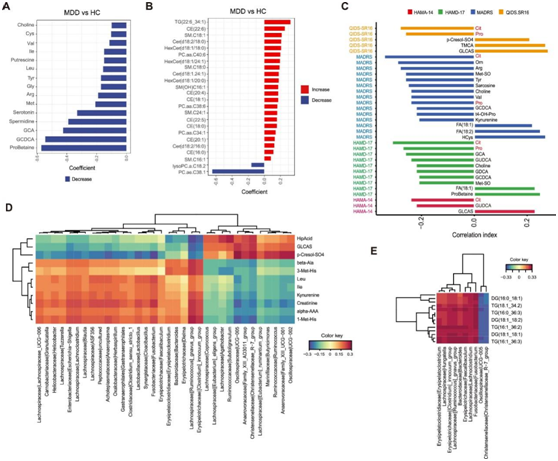
图2. MDD 血浆代谢物的变化。
(A) HC 和 MDD 之间显著差异表达的代谢物 (LC-MS) 的条形图。(B) HC 和 MDD 患者之间显著差异表达的代谢物 (FIA) 的条形图。根据年龄、性别、BMI 和吸烟情况调整的线性模型计算显著差异表达的代谢物。(C) 条形图显示 MDD 中抑郁评分(HAMA-14、HAMD-17、MADRS、QIDS-SR16)与代谢物 (LS-MS) 之间的相关性。(D) 热图显示正则化典型相关分析(rCCA)的代谢组 (LC-MS) 和微生物组数据特征之间的相关性。(E)热图显示 rCCA 的代谢组 (FIA) 和微生物组数据特征之间的相关性。
03
MDD患者血液免疫亚群的变化及其与代谢物的相关性
为了全面检查MDD患者外周血免疫细胞亚型的变化,他们使用细胞计数法对免疫亚群比例进行了分析。结果显示MDD组和HC组在许多免疫细胞亚型上存在显著差异。MDD患者CCR2+ CD8T细胞、Trem1+经典单核细胞、IgD+ 记忆B细胞和浆母细胞的比例较高,而Treg及其亚型(静息Treg、活化Treg、CCR4+Treg)、滤泡T细胞(Tfr)、γδ T细胞(GDT)、Th17细胞、幼稚CD4 T细胞和CD4 T细胞的水平较低(图3A)。
然后,他们进一步研究了血液免疫细胞亚型与抑郁症严重程度之间的相关性(图3B)。值得注意的是,CCR5+CD4T细胞与所有抑郁评分之间存在显著的负相关,表明它们的减少与MDD的进展有关。CD36+Tregs与QIDS-SR16、HAMD-17和HAMA-14呈负相关,提示其在抑郁严重程度中有潜在作用。IL6+ CD8T细胞和CD39+ Tregs与多重抑郁评分呈负相关,暗示它们与疾病有关。另一方面,少数免疫细胞亚型与抑郁评分呈正相关。例如,CD8T细胞和效应CD8T细胞与MADRS呈正相关,经典单核细胞和单核细胞髓源性抑制细胞(momdsc)与HAMD-17呈正相关,Th2细胞与hamd-14呈正相关。他们进一步分析了血液代谢物和免疫亚群之间的相关性,以深入了解它们在MDD发展中的潜在相互联系。使用正则化典型相关分析(rCCA),揭示了代谢物和免疫亚群之间的相关性,四种小分子代谢物显示与免疫亚群相关(图3C)。具体而言,次黄嘌呤、乳酸(Lac)和牛磺酸胆酸(TMCA)与IL5+CD4T细胞、TNFα+ CD4T细胞、TNFα+ B细胞、IL2+ DC、CD127+单核细胞和经典单核细胞呈正相关,而与Treg、CD4T细胞、Ki67+ CD4T细胞、Ki67+ T细胞、Tfh细胞、TGFβ+单核细胞、CD40L+ CD8T细胞和CD28+ CD8T细胞呈负相关。相反,胱氨酸表现出相反的相关性模式。TG与不同单核细胞亚型、CCR5+ CD8T细胞呈负相关,与Th1、GZMB+ CD4T细胞、IL4+ CD8T细胞、IL4+ B细胞、GZMB+NK细胞呈正相关(图3D)。这些相关性表明,这些代谢物和免疫亚群在MDD的发展过程中存在潜在的相互作用。
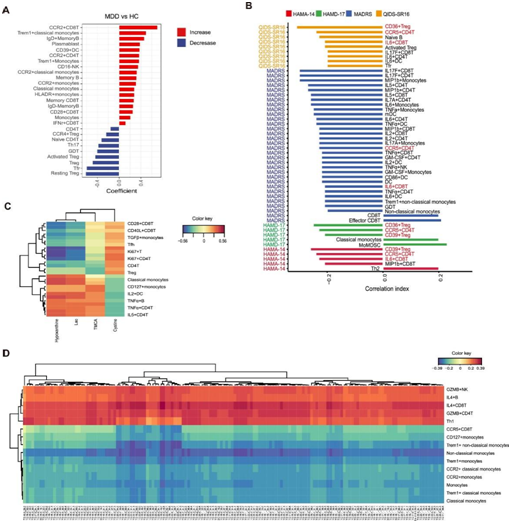
图3. MDD 中血液免疫细胞表型的改变。
(A) HC 和 MDD 之间显著差异的免疫细胞子集的条形图。根据年龄、性别、体重指数和吸烟情况调整的线性模型发现了显著差异的免疫细胞亚群。(B) 条形图显示 MDD 中抑郁评分(HAMA-14、HAMD-17、MADRS、QIDS-SR16)与免疫细胞亚群之间的相关性。(C) 热图显示 rCCA 的代谢组 (LC-MS) 和免疫细胞之间的相关性。(D) 热图显示 rCCA 代谢组 (FIA) 和免疫细胞之间的相关性。
04
MDD的免疫亚型及其与复发的关系
他们进一步利用基于主成分(HCPC)的层次聚类将MDD患者分为两种不同的免疫亚型,称为免疫亚型1和免疫亚型2(图4A)。免疫亚型1表现出较高水平的 Treg、T 细胞和 CD4 T 细胞,同时表现出较低水平的Trem1+ 经典单核细胞、经典单核细胞、单核细胞等。另一方面,免疫亚型 2 显示较高水平的 HLADR +单核细胞、Trem1 +经典单核细胞、经典单核细胞等,同时伴有较低水平的Treg、T 细胞和 CD4 T 细胞。
接下来,他们检测了外周免疫细胞亚型与抑郁症临床特征之间的相关性。有趣的是,他们发现免疫细胞亚型与 MDD 发作次数之间存在显著相关性,而与首次发病年龄、疾病持续时间或抑郁评分没有观察到显著相关性。特别值得注意的是免疫细胞亚型与 MDD 复发之间的高度相关性(图 4B),这表明外周免疫细胞亚型可能作为复发风险的预测标记。
然而,外周血特征出现显著差异。复发患者表现出较高比例的CD127 +单核细胞、CD4T和CD8T以及树突状细胞(DC)亚型,同时表现出较低比例的Treg亚型(图4C),这些发现表明复发患者的炎症状态加剧。在血液代谢物方面,他们观察到两组之间的差异。复发患者表现出较低水平的小分子代谢物,例如3-吲哚丙酸 (3-IPA)、腐胺、甘氨酸 (Gly)、精氨酸 (Arg)、瓜氨酸 (Cit) 和谷氨酰胺 (Gln),但表现出较高水平与首次发作患者相比,牛磺脱氧胆酸(TDCA)、蛋氨酸亚砜(Met-SO)和牛磺胆酸(TCA)的含量(图4D)。在血脂方面,复发患者的多个PC、Hex2Cer(d18:1/24:0)和Hex3Cer(d18:1/22:0)高于首次发病患者,而Cer(d 18:1/25:0)、Cer(d18:2/22:0)和HexCer(d18:2/24:0)低于首次发病患者(图4E)。此外,他们还分析了首次发病患者和复发患者肠道菌群的差异。复发患者中Hungatella、Anaerotruncus、Enterococcus等类群的丰度较高,Paraprevotella、Sellimonas、Dubosiella等类群的丰度较低(图4F)。
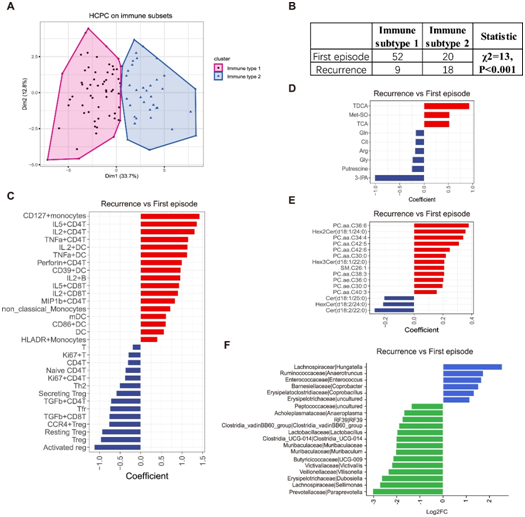
图4. MDD 的免疫亚型及其与复发的关联。
(A) 使用差异表达的免疫子集特征对 99 名受试者进行主成分 (HCPC) 分析的层次聚类得到的二维因子聚类图。(B) 免疫亚型与疾病发作之间的相关性。(C) 首次发病和复发之间显著差异的免疫细胞亚群的条形图。(D-E) 首次发病和复发之间显著差异代谢物的条形图。(F)比较首次发病组和复发组在属水平上的丰度差异。
05
鉴别MDD和HC的综合特征
通过DIABLO分析,他们观察到基于免疫细胞、代谢物和肠道微生物群的关键特征,MDD和HC样本之间存在明显的差异(图5A)。在免疫亚群方面,HC个体表现出更高水平的静息Treg,而MDD患者表现出更高水平的IgD+ 记忆B细胞、记忆B细胞、CCR2+CD8T细胞等。在细胞因子方面,HC样品显示亚精胺、蛋氨酸(Met)、酪氨酸(Tyr)等水平升高(图5B)。在肠道菌群方面,MDD样品显示出较高水平的Desulfovibrio、Dialister、Butyricimonas等,而HC样品显示出较高水平的Lactobacillus、Anaerotruncus、Granulicatella等。通过实施DIABLO整合分析,他们确定了不同组学水平上高度相关的特征(图5C)。具体来说,免疫亚群如活化的treg、CCR4+ treg、幼稚 CD4T细胞和静止的treg,与多种氨基酸,包括蛋氨酸(Met)、异亮氨酸(Ile)、苯丙氨酸(Phe)等,形成了一个亚群落。另一个亚群包括免疫亚群,如CD86+dc、dc、mdc和肠道微生物,包括厌氧voracaceae。Family_XIII_AD3011_group、Ruminococcus和Defluviitaleaceae_UCG.011。此外,代谢产物如鸟氨酸(Orn)、脯氨酸(Pro)、丙氨酸(Ala)和肠道微生物如clostridiides、Anaeroplasma和Lachnoclostridium在同一亚群落中发现,表明它们之间可能存在相互作用。
总的来说,这项综合分析提供了一组多组学特征,用于区分重度抑郁症患者和HC患者,并揭示了每个群体中变量之间的相关性。
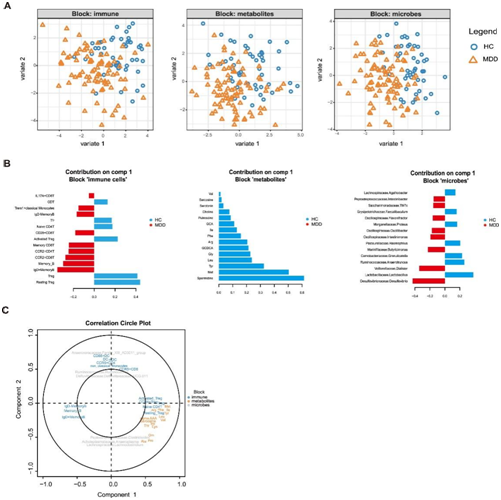
图5. 用于区分 MDD 患者和 HC 的综合特征。整合三个组学数据集:免疫细胞、代谢物和微生物。
(A) 每个组学数据集的分量 1 和 2 上的各个样本的散点图。(B) 每个组学数据集的组件 1 上前 15 个特征(按加载权重排名)的加载图。(C) 相关圆图。将每个特征与分量 1 和 2 之间的相关性绘制为散点图。
+ + + + + + + + + + +
结 论
本项研究对从包括 MDD 患者和健康对照的观察队列中获得的粪便和血液样本进行了多组学分析。肠道微生物群的 16S rRNA 测序显示 MDD 中的结构改变,其特征是肠球菌增加。肠道微生物群的宏基因组测序显示,MDD 存在显著的功能改变,包括乙醛酸循环超级途径的上调以及脂肪酸降解和各种代谢途径的下调。血浆代谢组学显示重度抑郁症患者的氨基酸和胆汁酸减少,同时鞘脂和胆固醇酯增加。值得注意的是,涉及精氨酸和脯氨酸代谢的代谢物减少,而鞘脂代谢途径增加。血液免疫细胞亚型的质谱流式分析显示,MDD 中促炎免疫亚群增加,抗炎免疫亚群下降。此外,本项研究结果揭示了 MDD 疾病严重程度相关的因素。有趣的是,他们将MDD分为两种与疾病复发高度相关的免疫亚型。此外,他们还建立了区分 MDD 和 HC 的区别特征。这些发现有助于全面了解MDD发病机制,并为生物标志物的发现提供宝贵的资源。
+ + + + +



