

 English
English文献解读|Cell(42.5):微生物移植导致的微生物组不匹配会导致持续的脱靶代谢和免疫调节效应
✦ +
+
论文ID
原名:Microbiome mismatches from microbiota transplants lead to persistent off-target metabolic and immunomodulatory effects
译名:微生物移植导致的微生物组不匹配会导致持续的脱靶代谢和免疫调节效应
期刊:Cell
影响因子:42.5
发表时间:2025.06.06
DOI号:10.1016/j.cell.2025.05.014
背 景
粪便微生物移植 (FMT) 是一种越来越常用的干预措施,用于促进肠道菌群失调的修复。虽然FMT对复发性艰难梭菌感染有效,但其在其他疾病(包括肥胖症、自闭症、癌症和炎症性肠病)中的机制、疗效和脱靶后果仍不清楚。由于小肠 (SB) 中以厌氧菌为主,FMT恢复局部肠道菌群(尤其是小肠)的适用性值得质疑。
实验设计

结 果
01

SB和大肠(LB)的微生物移植
研究团队分析了7名接受上消化道内镜FMT的受试者,并对FMT前和1个月后的样本进行了16S rRNA扩增子测序(图1 A)。虽然β多样性未发现显著差异(图1 B),但他们发现到FMT后严格厌氧菌有所增加(图1 C),SB中厌氧菌定植显著增加。
由于SBM和LBM之间的差异,他们假设SB的厌氧定植会对宿主产生不良后果。由于这在人体中难以研究,他们利用抗生素后不同菌群移植(MT)的模型来更好地分析区域菌群错配的后果。来自 Jackson 实验室 (JAX)的C57Bl6小鼠接受了为期 2 周的抗生素 (ABX)(包括氨苄西林、万古霉素、新霉素和甲硝唑)治疗,之后给它们进行单次小肠菌群(SBM) 移植(空肠和JMT [空肠微生物移植])、大肠菌群(LBM)移植(结肠FMT)、SBM 和 LBM 混合移植(盲肠和 CMT [盲肠微生物移植]) 或无 MT(磷酸盐缓冲溶液 [PBS]),这些移植来自内部饲养的供体,并通过口服管饲法递送(图 1D)。将 CMT 和 FMT 接种物稀释 1:10 以容纳 LB 中更高的 CFU。首先,他们通过 16S rRNA 测序分析每个区域肠道生态系统中的微生物群,以确定 MT 是否改变了组成。处死时,直接从每个肠道区域采集肠腔内容物,因此代表了LB和SB特有的微生物群落。比较整个肠道的β多样性表明,MT和区域(SB vs. LB)均存在分离(图1 E-F),这表明微生物群及其所处的生态系统的差异决定了区域微生物群的组成。对每个肠道区域的分析表明,每个肠道区域内都有独特的微生物组成以及MT依赖的α多样性变化。
接下来,他他们通过分析每个 MT 特有的扩增子序列变体 (ASV) 的百分比来评估移植微生物的植入情况。众所周知,不同动物设施之间的微生物群落差异很大,每个小鼠群落都含有不同的微生物菌株。在所研究的微生物群落中,明确的供体菌株占 ASV 的 5%–35% (图1 G)。不出所料,JMT 微生物在空肠中定植 (20%),在盲肠和结肠中的效率较低 (15%)。FMT 和 CMT 在盲肠和结肠的厌氧环境中定植率最高,为 28%–30%,在需氧空肠中的植入率较低 (5%–10%)。这对应于 JMT 后需氧菌水平较高,FMT 后厌氧菌水平较高(图1H-I)。这些包括JMT 小鼠中的需氧属Lactobacillus和 FMT 小鼠中的厌氧属Ileibacterium、Dubosiella、Faecalibaculum、Enterorhabdus和Desulfovibrio(图 1 J)。整个细菌家族似乎在给定的肠道区域内都不存在,证明了 MT 对恢复区域微生物群的巨大影响(图 1 K)。确定了在受体动物中检测到的供体移植材料中独特的非冗余 ASV 的数量。在供体移植材料中共鉴定出 449 个非冗余 ASV。在所有实验中,在 JMT 供体材料中检测到 109 个 ASV,在 FMT 中检测到 217 个,在 CMT 供体材料中检测到 422 个 ASV。在这些 ASV 中,JMT 受体小鼠平均检测到18.27 个, CMT 受体小鼠平均检测到34.42 个,FMT 受体小鼠平均检测到 27.97 个。植入和 MT 特异性成分的差异在长达 3 个月的观察期内均有存在,这强调了单次移植的持久植入(参见数据 S1)。为了分析 MT 后产生的微生物群落功能潜力的变化,他们对这些动物的结肠内容物进行了宏基因组测序,并进行了 KEGG 直系同源 (KO) 通路分析,比较了给定移植物与 PBS 对照中富集的通路(图 S3A)。通路分析表明,CMT 和 FMT 后恢复的通路(分别为 95 条和 79 条)比 JMT 后恢复的通路(45 条)要多,其中许多通路在所有移植中是共有 的,但也有一些是特定于其他移植的(图S3B-C)。有趣的是,JMT 宏基因组特别富集脂肪酸生物合成途径,而 β-氧化基因、胆汁盐水解酶和 7α-HSDH 的含量较低(图S3D-K)。
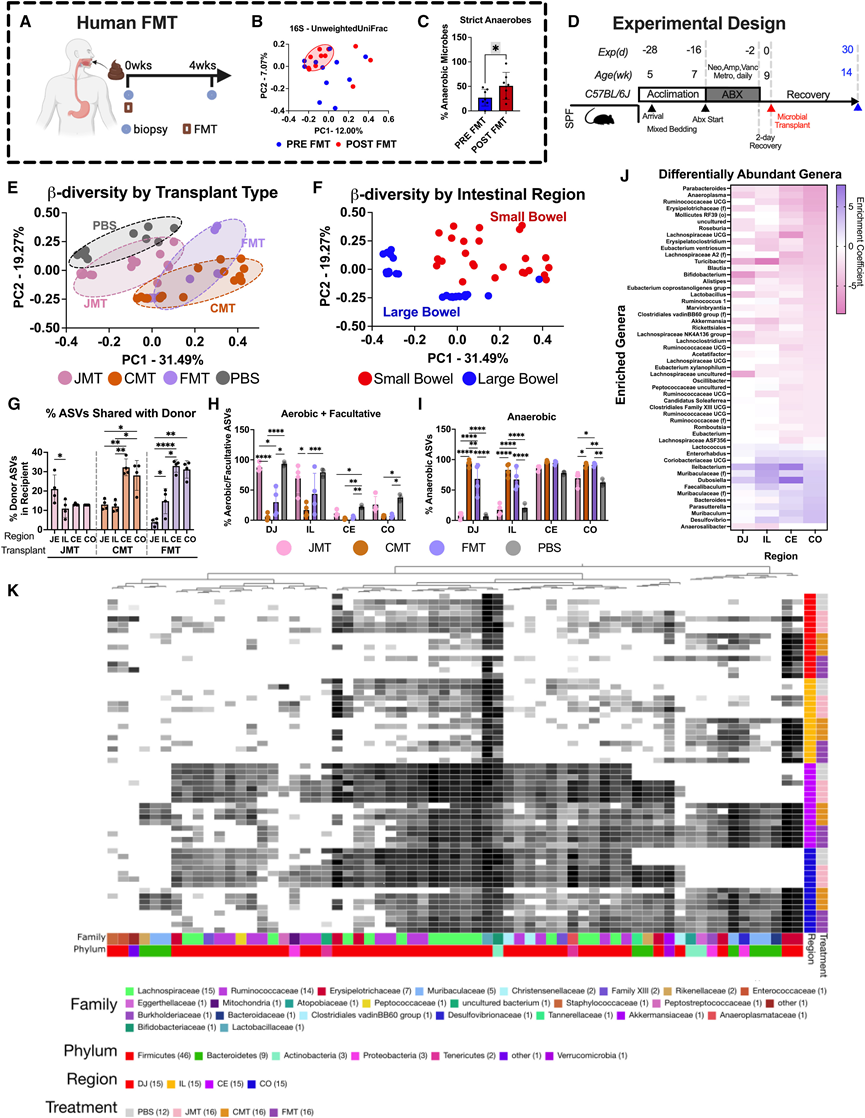
图1. 小肠和大肠微管在肠道内定植并改变本区域微生物群落。
(A) 人类 FMT 中的样本采集。(B) MT后1个月肠道内容物的16S rRNA测序。(C) FMT 前后严格厌氧菌的百分比。(D) FMT 前后严格厌氧菌的百分比。(E-F) MT 后 1 个月肠道内容物的 16S rRNA 测序。(G) 空肠、回肠、盲肠和结肠中每种移植类型中来自供体(MT 植入)的 ASV 的百分比。(H-I) 分别通过 16S rRNA 测序得出需氧兼性微生物和厌氧微生物的百分比。(J) JMT 和 FMT 在每个肠道区域中的差异丰度属。(K) 不同处理组和肠道区域的前 50 个属的热图。

图S3. 宏基因组测序揭示了功能潜力的差异。
(A) MT 后 1 个月对结肠内容物进行测序。(B) 恢复的功能模块的重叠表明,任何 MT 都会恢复大量的功能通路,但 FMT 和 CMT 恢复了结肠中 JMT 无法恢复的 42 条独特通路。(C) 通过 PBS 为每个 MT 恢复的显著通路显示为标准化富集分数。(D-K) KEGG 直系同源物 (KO) 的基因覆盖率映射到胆汁酸基因和脂质通路。(L) KEGG 直系同源物 (KO) 的基因覆盖率映射到胆汁酸基因和脂质途径。(M) 然后使用 SPARCC 将重建的宏基因组组装基因组 (MAG) 的丰度与肝脏转录组变化相关联,并绘制两个值以识别包含特定功能并与宿主转录变化相关并可能导致观察到的变化的 MAG。
02
SB 和 LB的微生物移植改变区域和系统代谢物
区域差异延伸到代谢物浓度,部分原因是由于地理上特定的微生物转化。这些代谢物可以作为信号分子来影响宿主的健康。为了表征微生物组的功能输出,他们对十二指肠空肠和结肠腔内容物进行了有针对性的代谢组学研究。
他们分析了 300 多种已知的人类和微生物产生或改造的化合物,包括 BA、SCFA、氨基酸和碳水化合物/糖。在十二指肠空肠或结肠中,JMT组和 FMT 组之间的 70 种化合物含量存在差异(图 2A-B)。使用主成分分析 (PCA) 可视化 JMT组和 FMT组代谢物之间的差异,突出 MT 类型对微生物组功能输出的影响(图 2C-E)。PBS组和 FMT 组显示出相似的代谢组学特征。在 FMT 后的十二指肠空肠中,他们检测到了较高水平的丙酸;较低水平的复合碳水化合物(如纤维二糖)和单糖(如蔗糖、葡萄糖和果糖);以及低糖醇,例如甘露醇、麦芽糖醇、山梨糖醇和半乳糖醇。与JMT相比,FMT结肠中检测到的糖/糖醇和氨基酸(包括亮氨酸、异亮氨酸、酪氨酸、蛋氨酸、缬氨酸、苯丙氨酸、天冬酰胺和赖氨酸)含量较低。他们观察到胆汁酸 (BA)具有较大的差异,包括较低的结合BA,例如牛磺鹅去氧胆酸(tauroCDCA)、牛磺胆酸(tauroCA)和甘氨胆酸(glycoCA)。他们发现 JMT 增加了十二指肠空肠中 BA 的总浓度(图 2 F),降低了结肠中次级 BA 的百分比(图 2 G),并且结合 BA 比率呈趋势但不显著增加(图 2 H)。
他们发现 MT 还会影响血浆中的全身循环代谢物,PCA 和热图均可显示 JMT 和 FMT 之间存在明显差异(图 2J-I)。血浆碳水化合物和氨基酸天冬氨酸、谷氨酸和脯氨酸在 JMT 中富集,而异亮氨酸在 FMT 中富集(图2I)。然而,发现循环脂肪酸棕榈酸、肉豆蔻酸、亚油酸和顺式油酸在 FMT 中的浓度高于 JMT。这与循环 BA 的变化相关(图2K-M)。JMT处理后,动物的血浆总 BA 浓度显著升高,并检测到结合 BA 增加。这些差异是由JMT小鼠体内β-鼠李糖酸和CA的富集所致,其代谢组变化持续长达3个月,其特征是氨基酸、生物素化酶和碳水化合物浓度的变化相似。总之,这些数据表明SBM和LBM会影响区域肠道菌群的组成和功能输出,从而影响多种由微生物修饰和产生的代谢物。
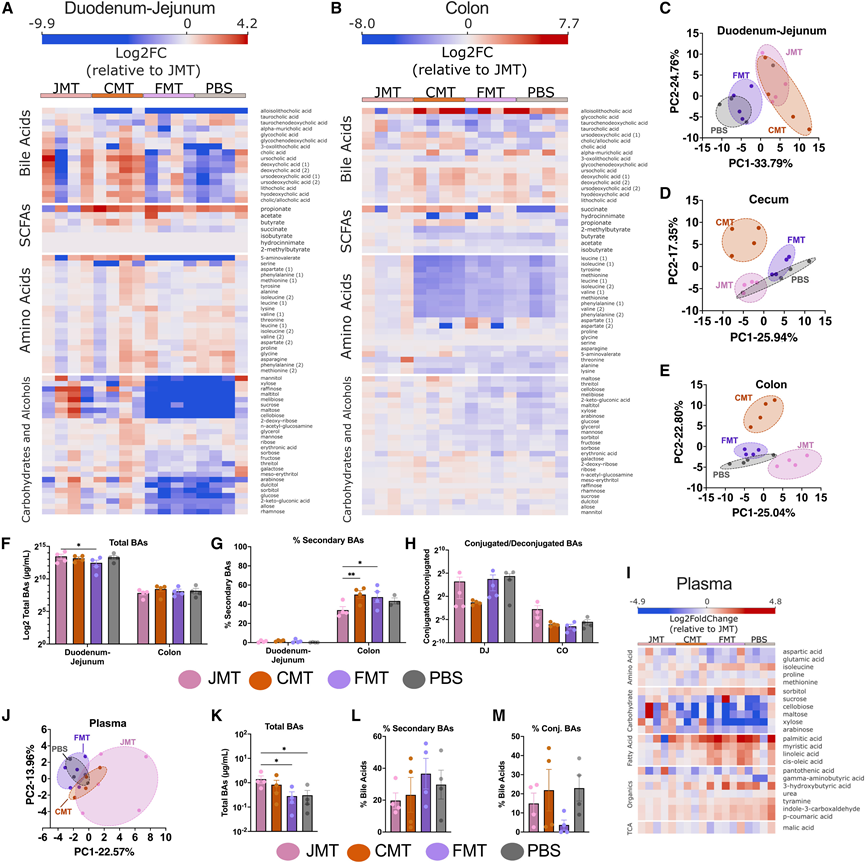
图2. 小肠和大肠微管改变区域和全身代谢物池。
(A-B) 靶向代谢组学中十二指肠-空肠和结肠内差异丰度代谢物的热图。(C-E) 十二指肠空肠、盲肠和结肠中标准化代谢物丰度的主成分分析(PCA)。(F-H) 对十二指肠空肠和结肠中检测到的 16 种最丰富的胆汁酸进行靶向代谢组学和绝对定量分析,以总胆汁酸、次级胆汁酸百分比和结合/去结合胆汁酸表示。(I) 血浆中差异丰度代谢物的热图。(J)血浆中代谢物的 PCA。(K-M) 对血浆中检测到的前16种胆汁酸进行靶向代谢组学分析和绝对定量分析,以总胆汁酸、次级胆汁酸百分比和结合胆汁酸百分比表示。
03
无菌(GF)小鼠与ABX模型有相似之处
为了探究SBM和LBM如何在没有竞争、抗生素作用和自身接种(通过食粪)粪便菌群的情况下定植,他们将JMT和FMT移植到无菌(GF)小鼠(这些小鼠只能将移植的菌群重新接种到自身)体内,并进行了16S rRNA扩增子测序和代谢组学分析。
与抗生素后小鼠模型类似,他们发现移植类型和肠道区域影响微生物组成(图 3 A-B)。除回肠外,每个肠道区段都有不同的群落。虽然没有竞争微生物,FMT 在 SB 中的定植效率不如 LB(高出约 20%)(图 3 C)。虽然 JMT 微生物在 SB 中的定植效率高于 FMT 微生物(约 40%),但 JMT 在 LB 中的定植有所不同(20%-90%)。JMT 后 SB 需氧和兼性定植增加,FMT 后厌氧定植增加(图3D-E)。然而,在LB中没有发现差异。有趣的是,与JMT-GF小鼠相比,JMT-SPF(无特定病原体)小鼠在SB中表现出更高水平的需氧菌和更低水平的厌氧菌,这可能是由于GF小鼠SB中氧气的高度可变性(图3F-G)。差异富集的微生物与SPF小鼠相似,包括JMT富集的需氧乳酸杆菌和FMT富集的Rumminococcaceae、Eubacterium、Coriobacteriaceae、Faecalibaculum和Enterorhabdus(图3H)。这些与 SPF 动物中发现的代谢物概况相关,包括富含 JMT 的肠道氨基酸、SCFA 减少、十二指肠空肠和结肠中总 BA 增加、结肠中结合 BA 增加以及次级 BA 减少。总之,这些数据表明,在没有抗生素并发症、原生微生物群竞争和粪便微生物群重新接种的情况下,GF 小鼠在整个肠道中显示出相似的定植和代谢物概况模式,包括 SB 的厌氧定植。此外,GF 小鼠表现出改变的区域和全身 BA ,这与施用抗生素后小鼠和已知可防止食粪的“粪便收集”杯模型一致,包括总BA增加和次级BA减少。
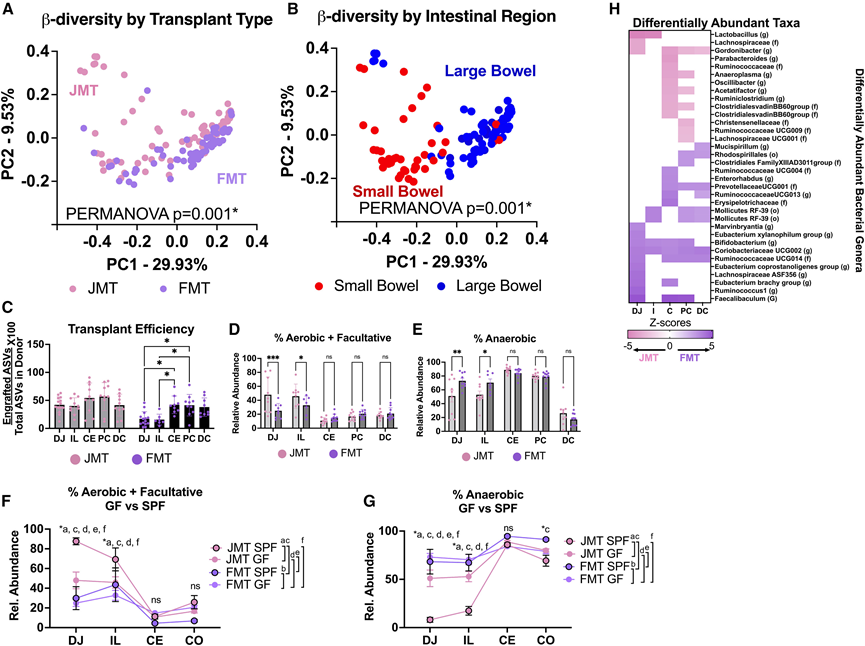
图3. GF小鼠与ABX模型相似。
(A-B) MT 后 1 个月肠道内容物的 16S rRNA 测序。(C) MT 转移效率,计算为来自 MT 的 ASV 总数/MT 中 ASV 总数的百分比。(D) SB 中 JMT 的需氧 + 兼性微生物的百分比较高。(E) FMT 后 SB 中的厌氧微生物百分比较高。(F-G) 需氧+兼氧或厌氧微生物的 SPF 和 GF 丰度比较。(H) 线性判别分析效应大小(LEfSe)用于检测每个区域中差异丰富的微生物。
04
微生物移驱动不同的肝脏转录程序,这些程序可持续长达 3 个月
许多活性炭、碳水化合物和氨基酸都具有生物活性,作为信号分子对代谢和总营养通量有重大影响。来自肠道的代谢物在整个肠道中吸收,由肠道血管系统收集,并通过肝门静脉进入肝脏。肝脏是人体的主要代谢器官,也是肠道外第一个接触微生物产物的器官。鉴于肠道定植、代谢物库的变化以及豆粕在代谢中的作用,他们探究了区域性微生物失配如何影响肝脏等远处器官。
他们进行了转录组分析(RNA-seq)以探究MT后的肝脏转录谱。每个MT组的肝脏转录组都是独特的,JMT和FMT接受者之间的差异最大,而CMT接受者表现出中间表型(图4 A)。差异表达基因(DEG)分析证实了JMT和FMT之间最大的差异,共有3928个DEG(图4 B)。在JMT与CMT组中发现了2504个DEG,在FMT与CMT组中发现了1805个DEG(图4C-D)。引人注目的是,JMT 富含代谢和脂质相关基因,包括含 ELMO 结构域 3 (Elmod3)、细胞色素 p450 Cyp4a14 和 Cyp4a32、周脂素 5 (Plin5)、过氧化物酶体生物合成因子 11 alpha (Pex11a)、溶质载体Slc25a47和酰基辅酶 A 硫酯酶 (Acot) 1/2。FMT 表达富集血清淀粉样蛋白 A3 (Saa3)、脂质运载蛋白 2 (Lcn2)、肝配蛋白 A3 (Efna3)、白细胞介素 33 (Il-33)、T 细胞受体相关跨膜适配器 1 (Trat1) 和 α 激酶 1 (Alpk1),这些基因均参与免疫功能。通路分析显示,14 条 FMT 富集的 KEGG 通路中有 10 条与免疫相关,包括“致病性大肠杆菌感染”、“细胞因子-细胞因子信号传导”、“趋化因子信号传导”、“RIG-I 样信号传导”、“TLR 信号传导”和“NOD 样信号传导”。相反,JMT中富集的顶级通路与代谢相关,包括“PPAR 信号传导”、“氧化磷酸化”、“脂肪酸代谢”、“不饱和脂肪酸合成”、“BCAA 降解”和“亚油酸代谢”(图 4 E)。移植后 3 个月检测到了转录变化,并在 GF 小鼠中观察到(图 4 F),这表明肝脏转录组的变化是持久的,并且是微生物移植的直接影响。
JMT 小鼠在 1 个月后体重最重,体重增加最多,并且能量消耗最低(图 4G-K)且活动量最低(图 4 J)。FMT 小鼠的食物摄入量是所有组中最低的,但与 JMT 小鼠相比,其能量消耗水平更高(图 4 I)。有趣的是,活动、食物摄入、体重维持和体重变化以及能量消耗表明 CMT 小鼠的代谢最活跃(图 4 G-K)。CMT 小鼠的低呼吸交换率 (RER) 表明脂质氧化增加,与能量消耗增加和脂肪作为能量来源的利用相一致(图 4 L)。
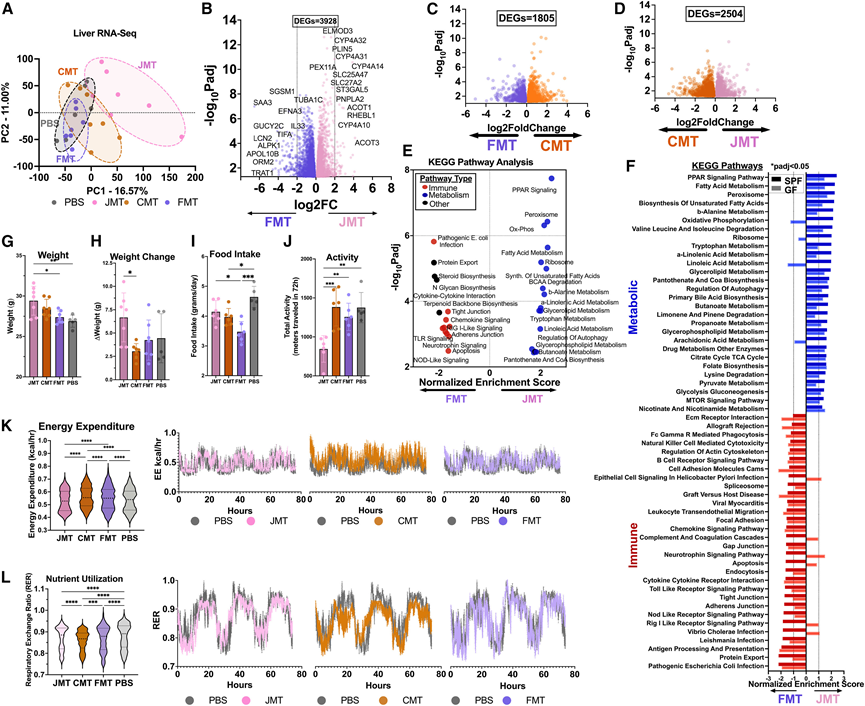
图4. 微管驱动不同的肝脏转录程序,这些程序持续存在并改变能量平衡。
(A) MT 后 1 个月肝组织的 RNA-seq。(B-D) JMT 与 FMT、CMT 与 FMT 以及 JMT 与 CMT 之间差异表达基因的火山图。(E)代谢通路分析。(F) GF 和 SPF 差异富集途径的比较。(G-J) 使用 Sable Systems Promethion 呼吸测量系统进行的能量平衡评估表明 JMT 小鼠与 FMT 小鼠在体重、体重增加、食物摄入量和活动方面存在差异。(K-L) 与 PBS 组相比,所有处理组的能量消耗和营养利用均存在差异。
05
小肠菌群(SBM)和大肠菌群 (LBM)改变肠道生态系统
显然,移植本区域和非本区域微生物会导致微生物组组成和功能发生剧烈变化,从而影响宿主的代谢和远端器官(即肝脏)。结肠和空肠转录组的 PCA 显示 87% 的变异(PC1),突显了基因表达之间的巨大差异(图 5 A)。他们注意到 FMT 小鼠的空肠比结肠样本的 PC1 差异较小,表明与结肠组织相似(约 20%)。为了更深入地探究这一点,他们检测了空肠和结肠标记基因的表达。与 FMT 和 PBS 对照组相比,JMT 受体小鼠的空肠粘膜中空肠特异性基因更加丰富(图 5 B)。这些包括Gata4和Gata6(GATA 结合蛋白),它们是空肠身份的中心转录调节因子;脂质结合和转运基因,包括载脂蛋白Apoa1、Apoa4和分化簇 36/脂肪酸转位酶(Cd36 / Fat);以及糖转运蛋白/溶质载体家族成员Slc2a2、Slc2a5和Slc5a1。JMT表达了更高水平的大多数糖转运蛋白。在结肠中,FMT 表达了更高水平的结肠特异性基因,包括驱动结肠身份的Satb2;钙激活氯通道调节剂 1 (Clca1)、碳酸酐酶 1 (Car1)、淋巴细胞抗原 6 家族成员 G (Ly6g) 和桥粒芯蛋白 2 (Dsg2);结肠特异性水转运蛋白水通道蛋白 8 (Aqp8);以及 SCFA 转运蛋白Slc16a3和Slc5a8(图 5C)。有趣的是,FMT 后通常在空肠中表达的抗菌肽富集,包括防御素 α 3 (Defa3)、溶菌酶Lyz1、Lyz2和基质金属蛋白酶 (Mmp7),并且与 Erb-B2 受体酪氨酸激酶 3 (Erbb3) 和无调性同源物 1 (Atoh1)、Paneth细胞发育基因。
这些数据表明,错配的非天然微生物可以重新编程组织的特性,从而增强有利于适应和植入的基因。这可以解释单次 FMT 后 3 个月空肠中厌氧菌的持续存在。为了定量 JMT 和 FMT 对空肠和结肠区域生态系统的改变程度,他们检测了来自健康、未经 ABX 治疗的 C57Bl6 小鼠的空肠和结肠黏膜刮片的 RNA-seq数据,并确定了排名前 1000 个的 DEG(图 5D-E)。基于这些基因,他们创建了“空肠标记”和“结肠标记”基因集,以对 MT 肠道组织进行富集分析。他们发现 JMT 富集了空肠特征,而 FMT 增强了空肠中的结肠特征(图 5 F)。在结肠中也观察到了类似的模式,其中 JMT 富集了空肠特征,而 FMT 增强了结肠特征(图 5 G)。JMT 在空肠中对驱动空肠特征具有更大的作用,而 FMT 在结肠中更好地驱动了结肠特征。这些基因的可视化分别证实了 JMT 和 FMT 之后空肠和结肠程序的增强。
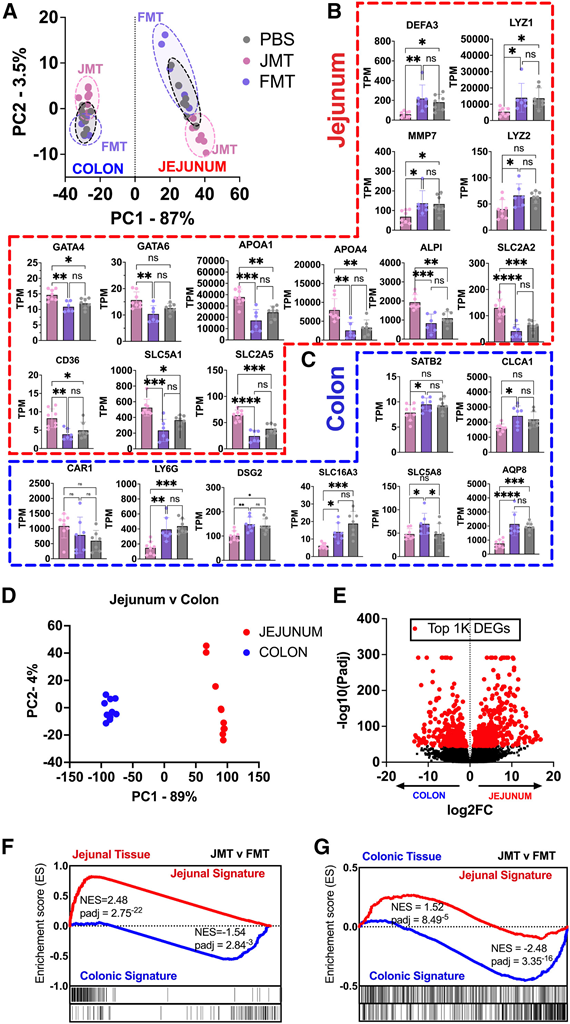
图5. 小肠和大肠微生物群使其区域生态系统分析。
(A) MT 后 1 个月肠黏膜组织的 RNA-seq。(B-C) 空肠中空肠标记基因和结肠中结肠标记基因的表达以每百万转录本数 (TPM) 表示。(D-E) 对健康、未经 ABX 处理的 C57Bl6 的空肠和结肠转录组进行 PCA 分析。(F) 空肠组织的通路分析表明,JMT 后空肠特征富集,FMT 后结肠中结肠特征富集。(G) 结肠组织通路分析显示,JMT后空肠信号富集,FMT后结肠信号富集。
06
人类 FMT 还会导致小肠上部持续存在结肠厌氧菌
将体外培养的原代人空肠活检样本分别用来自单个空肠造口供体的 10% 脱细胞人空肠内容物制剂或粪便浆液作为 JMT 和 FMT 的类似物(图 6 A)。虽然没有观察到在体内模型中观察到的所有标记身份转换的基因特征,并且在人类中这些基因并不严格地属于空肠(嗅球素 [OLFM4];脂肪酸结合蛋白 6 [ FABP6 ];APOA4;和碱性磷酸酶肠 [APLI])或结肠(SATB2;尾型同源框 2 [CDX2];碳酸酐酶 1 [CA1];和乳过氧化物酶 [LPO]),但与鼠模型相当的身份信号分别在 JMT 和 FMT 治疗后 24 小时内得到增强,这表明空肠到结肠之间的微生物介导的转变不是立即发生的(图 6B)。通路分析显示,脂质吸收、运输、生物合成和储存途径以及碳水化合物生物合成和利用途径在 JMT 处理的肠道中富集(图 6C)。然而,与脂质和碳水化合物代谢相关的通路在 FMT 处理的肠道中下调。重要的是,“脂质生物合成途径”在 JMT 处理的肠道中富集,而在 FMT 处理的肠道中下调(图 6C),这反映了 SB 微生物群增强脂质、碳水化合物和其他代谢过程的能力。虽然需要进一步研究来评估对特定人类身份标记的影响,但这些数据验证了从体内小鼠模型中得出的结论。
十二指肠转录组分析显示 FMT 前后样本之间没有统计学差异(图 6 D)。然而,他们发现十二指肠转录组的变化与厌氧菌定植水平的增加相关,这表明个体间反应取决于 FMT 植入(图 6 E)。他们观察到SATB 2表达增加和183 个上调结肠基因的结肠特征富集(图 6F-H)。通路分析显示十二指肠内耗氧过程增加,包括线粒体表达、氧化磷酸化和有氧呼吸(图 6 I)。小肠内耗氧量的增加可降低管腔内氧水平并支持更厌氧的环境,从而允许厌氧定植。总之,微生物能够改变粘膜生态系统以适应其原生环境的特征,并且这些过程可以在人类中发生。
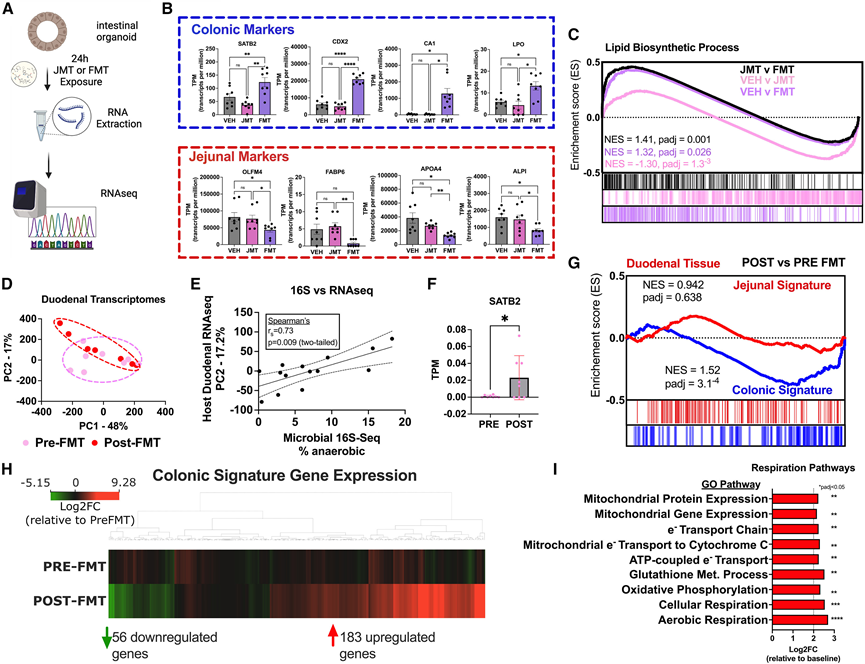
图6. 人体组织反映代谢和区域生态系统的变化。
(A) 将原代人类空肠活检样本在体外培养以产生空肠肠类物质,用人类 JMT(从空肠造口袋收集)或 FMT(从升结肠抽吸物)的无细胞制剂处理 24 小时,提取 RNA-seq。(B) FMT 富集结肠标记基因SATB2、CDX2、CA1和LPO。(C)基因富集排序列表。(D) 通过上消化道内镜进行人体FMT,并将其送至十二指肠末端。在随访内镜检查中,对FMT前和FMT后4周采集的十二指肠活检样本进行RNA测序和16S rRNA测序。(E) FMT 后十二指肠粘膜中定植的厌氧菌相对丰度增加,并与十二指肠转录组的变化相关。(F) FMT 前后十二指肠活检中的Satb2表达以每百万转录本数 (TPM) 显示。(G-H) 结肠特征基因样本的基因集富集分析显示 FMT 后富集,富集了 183 个结肠基因。(I) GO分析表明,FMT后线粒体、氧化磷酸化和有氧呼吸通路均富集。
+ + + + + + + + + + +
结 论
本项研究结果表明,FMT 期间可能出现区域性微生物植入不匹配,并对宿主健康和代谢产生负面影响,从而改变区域肠道生态系统的根本性质和特性。这些数据有力地支持了需要更客观的指标来评估 FMT 的短期和长期后果,并可能为未来基于微生物组的干预措施提供参考,使其能够同时考虑 SB 和 LB 环境及其中的微生物。此外,应进一步研究 FMT 的最佳给药途径,无论是经口给药还是上消化道内镜给药(而非结肠镜)。FMT后,暴露于代谢物的人类肠道类固醇和十二指肠活检样本的RNA-seq证实了小鼠的转录变化。因此,FMT后区域微生物组错配可能导致意想不到的后果,并需要重新考虑基于微生物组的干预措施。
+ + + + +



