

 English
English文献解读|Nature(64.8):一个独特的具核梭杆菌进化枝在结直肠癌领域占据主导地位
✦ +
+
论文ID
原名:A distinct Fusobacterium nucleatum clade dominates the colorectal cancer niche
译名:一个独特的具核梭杆菌进化枝在结直肠癌领域占据主导地位
期刊:Nature
影响因子:64.8
发表时间:2024.03.20
DOI号:10.1038/s41586-024-07182-w
背 景
具核梭杆菌(Fn) 是一种存在于人类口腔中的细菌,在健康个体的下胃肠道中很少发现,但在人类结直肠癌 (CRC) 肿瘤中含量丰富。癌症微生物组研究的一个关键挑战是超越肿瘤中微生物组成的表征,转向功能研究,目前尚不确定这些微生物引起疾病的确切机制。
实验设计
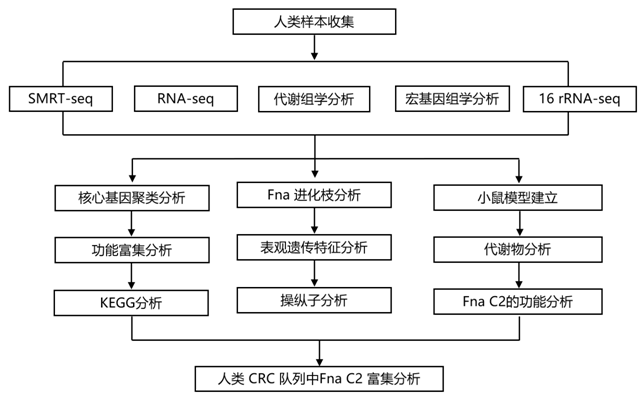
结 果
01

生态位富集的Fn基因和亚种
研究团队对 130 个人类 CRC 肿瘤进行了梭杆菌靶向培养,从 59 名独特患者中获得了 65 株与 CRC梭杆菌相关的菌株。鉴于Fn主要是一种口腔病原体,他们纳入了从非癌症个体口腔中分离出的81 株梭杆菌菌株,作为对照组。使用长读长单分子实时测序(SMRT-seq),他们为 146 种独特的梭杆菌菌株生成了完整且封闭的基因组,以及相应的表观遗传甲基化组(图1a),其中92%属于Fn(图1b)。由于Fn是CRC肿瘤中最常检测到的物种,因此他们将分析重点放在55个CRC相关和80个口腔相关的Fn基因组的比较上。
鉴于人类 CRC 肿瘤中的Fn菌株预计起源于人类口腔,但在无癌症个体的下胃肠 (GI) 道微生物群中却是罕见成员,他们推断与 CRC 相关的Fn菌株具有额外的遗传库以促进它们在人类结直肠癌肿瘤中的定植。他们根据生态位进行子集化,发现与口腔相关的Fn菌株相比,CRC 相关 Fn 菌株具有更小的辅助基因组(图1c)。功能富集分析发现CRC和口腔菌株中分别有483和241个基因聚类显著富集(图1d)。KEGG对724个基因聚类的同源分析显示,定位的基因聚类(31.2%)主要涉及假定的代谢功能和途径(图1e)。由于四个Fn亚种之间存在足够的遗传异质性,因此他们提出了将其重新分类为单独的种,将分析的分辨率提高到亚种水平[Fn亚种(Fna)、Fn亚种nucleatum (Fnn)、Fn亚种polymorphum (Fnp)和Fn亚种vincentii (Fnv)]。通过对Fn亚种的生态位比例分析发现,在这些Fn亚种中,只有Fna与CRC生态位显著相关,验证了之前的研究,而Fnn在口腔生态位中显著富集(图1f)。
他们观察到Fna菌株的一个子集缺乏fap2、cmpA和Fusolisin,并且通过rpoB基因分析显示,这些Fna菌株似乎形成了一个独特的Fna进化枝(图1g)。
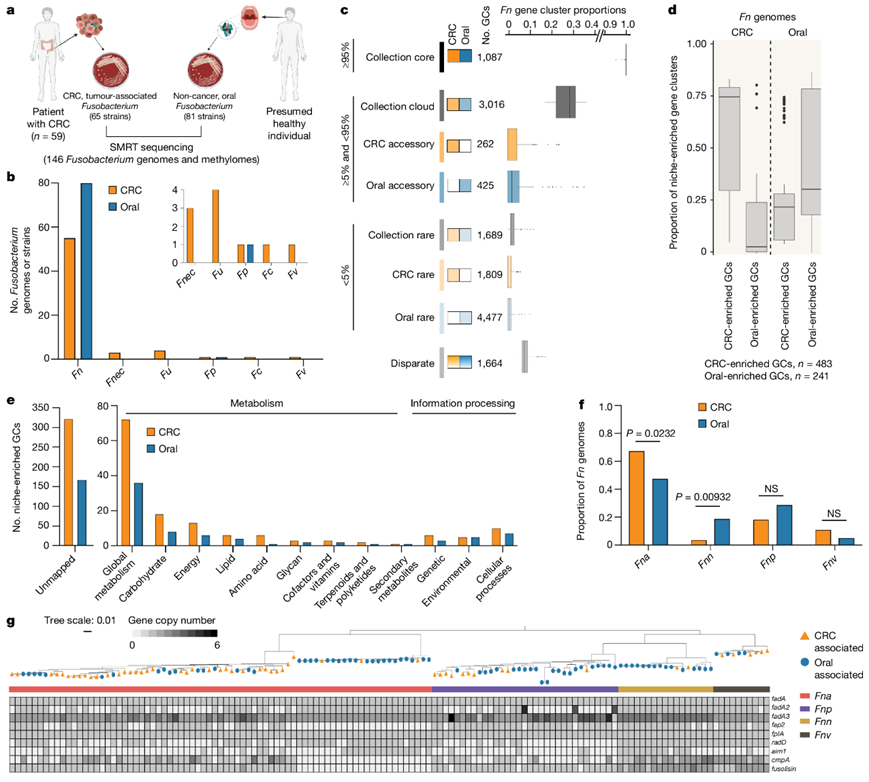
图1. Fn生态位的特征。
(a) 梭杆菌菌株收集示意图和独特菌株的测序策略。 (b) 柱状图显示了CRC(橙色)和口腔(蓝色)生态位内梭杆菌基因组的比例。(c) 按生态位划分的Fn全基因组子集的组成。 (d) CRC 相关和口腔相关Fn基因组中生态位富集基因聚类的比例。 (e)生态位富集基因聚类的直系同源物分析。 (f) 柱形图显示CRC 和口腔生态位内按亚种分组的Fn基因组的比例。(g) 典型Fn毒力因子的基因存在-缺失热图。
02
CRC 生态位中丰富的Fna分支
为了进一步检验Fna菌株形成两个不同分支的观察结果,他们比较了先前用于Fn 亚种分型的管家基因的系统发育树。对这些单标记基因的分析支持了存在两个不同的Fna 进化枝的观察结果,他们将其称为Fna进化枝 1 (Fna C1) 和Fna进化枝 2 (Fna C2)(图2a,图S2a)。为了量化Fna C1 与Fna C2的相关性,他们将平均核苷酸同一性 (ANI)(一种衡量基因组之间相似性百分比的成熟指数)与既定的 95% 物种阈值进行了比较。Fna分支之间的ANI 范围为 91.61% 至 93.11%,与其他Fn亚种之间的 ANI 相当(图2b)。此外,他们使用基因组图谱(GiG-map)工具可视化Fna基因组中存在的蛋白质编码基因的模式,并发现Fna C1和Fna C2具有不同的蛋白质编码基因(图2c)。主成分分析 (PCA) 进一步证明了这一点(图2d)。值得注意的是,常用的Fna 7_1 菌株组中含有Fna C2(图S2b)。
因此,他们重新评估了Fna作为两个遗传上不同的分支的遗传、表观遗传和生态特性。Fna进化支泛基因组的比较显示,Fna C1和Fna C2具有相似的核心基因组大小,但是Fna C2具有更大的辅助基因组(图S2c-d),这表明Fna C2菌株含有可能在结直肠癌肿瘤定植过程中有益的额外遗传因子。与此一致的是,个体基因组大小和含量的比较表明,与Fna C1菌株相比,Fna C2菌株具有更大的染色体大小(图S2e),更多的染色体外质粒以及更多的先天遗传防御和可移动遗传元件(图S2f)。Fna甲基组的PCA分析表明,Fna C1和Fna C2在表观遗传上也是不同的(图2e)。影响这种表观遗传分化的甲基修饰DNA基元是GTNm6AC (100% Fna C1, 0% Fna C2)、GCm6AG (100% Fna C1, 0% Fna C2)和Gm6ANTC (0% Fna C1, 63% Fna C2)(图2e)。尽管两种Fna分支均存在于口腔中且无显著差异,但只有Fna C2与CRC生态位显著相关(图2f)。他们进一步对来自结直肠癌患者配对肿瘤组织和唾液样本的公开可用16S rRNA基因测序数据进行了验证。与Fna C1相比,Fna C2在肿瘤样本中显著富集(图S2g)。然而,配对口腔样本中Fna分支之间没有统计学差异,这表明尽管两种Fna分支都存在于CRC患者的口腔中,但只有Fna C2在肿瘤生态位中富集(图S2g)。
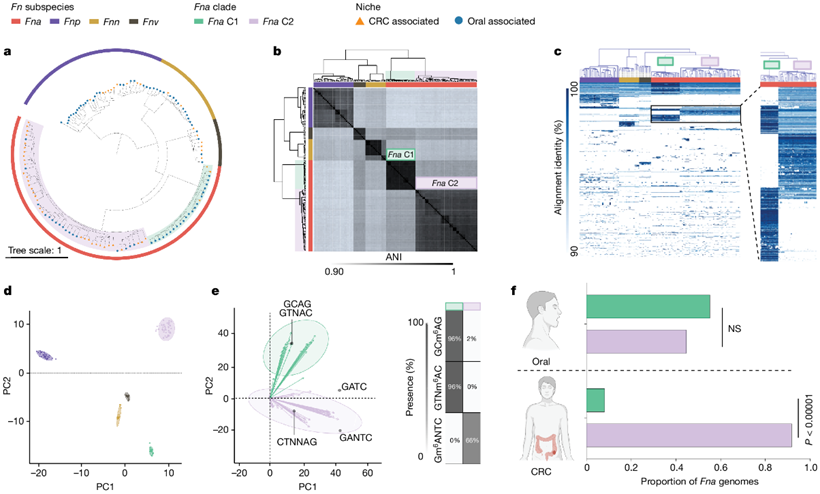
图2. Fna进化枝的遗传和表观遗传特征。
(a)最大似然全基因组系统发育树。 (b) ANI 矩阵的聚类树状图。(c) Fn基因组中蛋白质编码基因的 GiG-map 可视化。 (d)主成分分析(PCA)。 (e) 左:Fna全基因组甲基修饰核苷酸序列的 PCA。右:表格显示Fna进化枝中每个基序的分布。(f) 柱形图显示Fna CRC 相关基因组和Fna口腔相关基因组的比例。
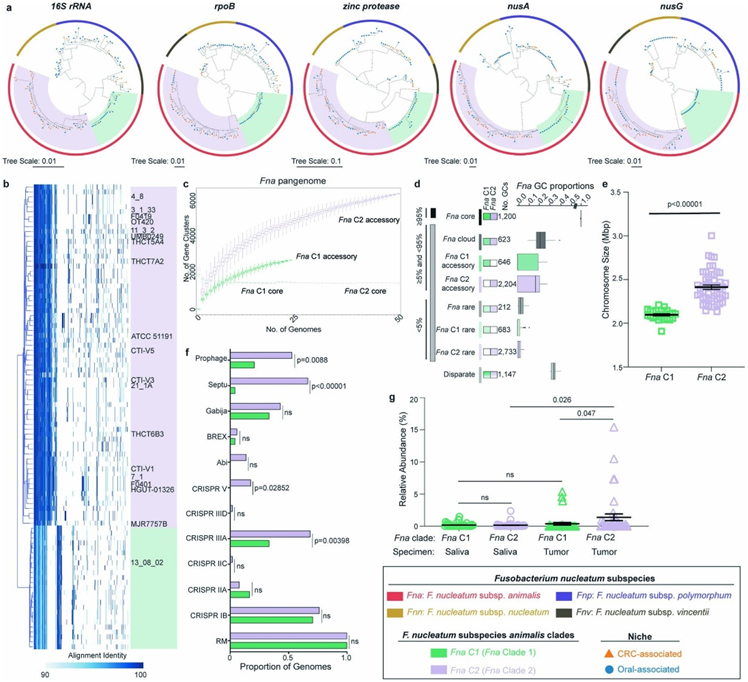
图S2. Fna进化枝的系统发育学和基因组学。
(a) Fn单标记基因、16S rRNA、rpoB、锌蛋白酶、nusA和nusG的最大似然系统发育树。(b) Fna基因组中蛋白质编码基因内容的GiG-map可视化。(c) 按Fna进化枝、Fna C1(绿色)和Fna C2(淡紫色)划分的Fna全基因组大小,并标记了各自的核心和辅助基因组。(d) 按进化枝划分的Fna全基因组子集的组成。 (e) 柱形图表示Fna C1 和Fna C2中的染色体大小。 (f) 柱形图显示包含先天细菌遗传防御系统子集的Fna基因组的比例。 (g) 图表显示来自 39 名结肠直肠腺癌患者的配对唾液(圆形)或肿瘤活检(三角形)样本中Fna C1(绿色)和Fna C2(淡紫色)的相对丰度百分比。
03
人下消化道Fna C2的富集
全基因组分析显示,Fna由两个不同的进化枝组成,但只有Fna C2 在 CRC 生态位中富集。该生态位中Fna C1水平较低可能是由于肿瘤定植潜力较差或者Fna C1 无法逃避免疫清除。为了探究这些可能性并揭示Fna进化枝特异性遗传因素,他们对所有 75 个Fna基因组(24 个Fna C1、51 个Fna C2)进行了全面的Fna进化枝间比较分析。与Fna C2 相比,Fna C1 中的典型Fn毒力因子(包括由radD、aim1和fadA2编码的粘附素)显著富集(图1g,图3a),表明它们在口腔中的重要作用。
每个进化枝的Fna菌株与人结肠癌细胞系 (HCT116) 的共培养表明,与Fna C1 菌株相比, Fna C2 具有显著更高水平的癌症上皮细胞侵袭能力(图3b),表明个体菌株的差异入侵潜力和空气耐受性。此外,Fna进化枝在形态上不同,Fna C2 细胞比 Fna C1细胞明显更长。
他们推断,Fna分支独特基因组含量的描绘可以揭示迄今为止未知的遗传因素,使Fna C2转运到人类结肠生态位并在其中存活。功能相关的细菌基因通常形成共同调控单位(操纵子),他们应用了泛基因组方法(PPanGGOLiN)来评估Fna进化枝独特的遗传因素是否形成推定的操纵子(图3c)。PPanGGOLiN 分析表明Fna C2 同线性模块主要与代谢机制相关。因此,Fna C2 对 CRC 生态位的病理适应是多因素的,除了典型的Fn毒力因子之外,增强的代谢能力还可能促进 Fn C2 的病理适应。
接下来,他们重点研究了两个与Fna c2相关的假定操纵子,这些操纵子与乙醇胺(EA)代谢(eut)和1,2-丙二醇(1,2- pd)代谢(pdu)有关(图3d)。
对CRC 患者 (n = 627) 和健康个体 (n = 619) 队列的粪便宏基因组数据集的分析表明,eut和pdu操纵子在 CRC 患者中显著富集(图S4a)。他们评估了Fna细胞暴露于这些肠道相关代谢物后的整体转录组反应。暴露于 EA 或 1,2-PD 后,对代表性Fna C1 和Fna C2 菌株进行转录组分析(RNA-seq)表明,这两个Fna分支均出现显著的转录组变化(图S4b-d)。由于Fna C1 缺乏eut和pdu,他们推断Fna C1 中暴露于 EA 或 1,2-PD 后的显著转录组变化将独立于这些操纵子。因此,他们重点关注 Fna C2 细胞中差异表达的基因。结果表明,在Fna C2中,eut和pdu基因分别在EA和1,2- pd的作用下发生转录上调(图3e-f)。
此外,暴露于EA或1,2- pd的Fna C2细胞显著上调了13.02%的Fna C2相关基因,包括典型的Fn毒力因子。虽然在这两个分支中都存在,但radD和aim1在Fna C2而非Fna C1细胞中存在EA时上调。当Fna C2细胞暴露于EA(cmpA、fusolisin和fap2)或1,2- pd (fap2)时,Fna C2中特有的毒力因子也会上调(图3e-f)。Fna C2相关基因和毒力因子的上调是与人类上皮细胞相互作用的重要因素,这表明,在它们转运到人类胃肠道后,对这些分子的感知可以诱导与口外生态位适应一致的Fna C2转录谱。
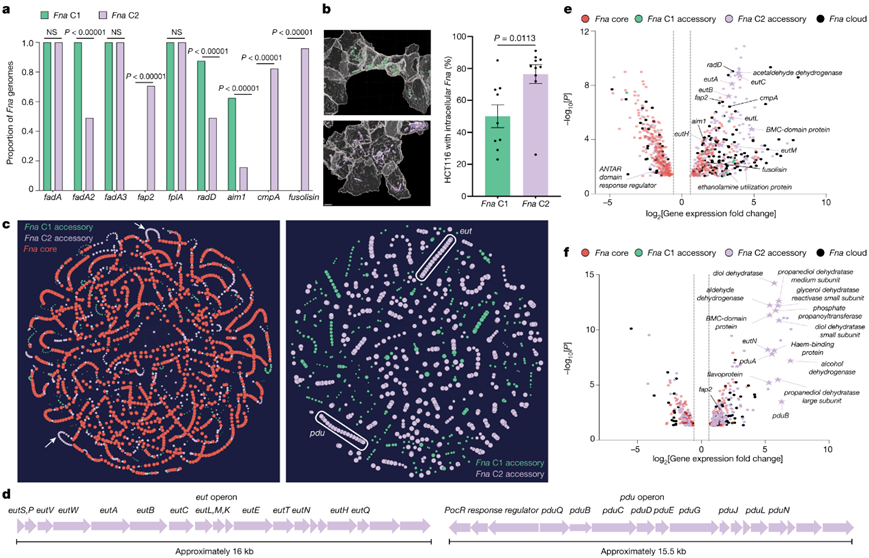
图3. Fna进化枝间比较分析。
(a) 基因存在与不存在的柱形图,描绘了包含典型Fn毒力因子的Fna基因组的比例。(b) 左:对与代表性Fna C1(绿色)或Fna C2(淡紫色)菌株共孵育的结肠癌上皮细胞(HCT116;灰色)进行计算共聚焦分析。右:显示具有细胞内Fna的 HCT116 细胞百分比的条形图。 (c) Fna全基因组的PPanGGOLiN图谱。(d) Fna C2 操纵子的示意图。(e-f)差异表达基因分析。
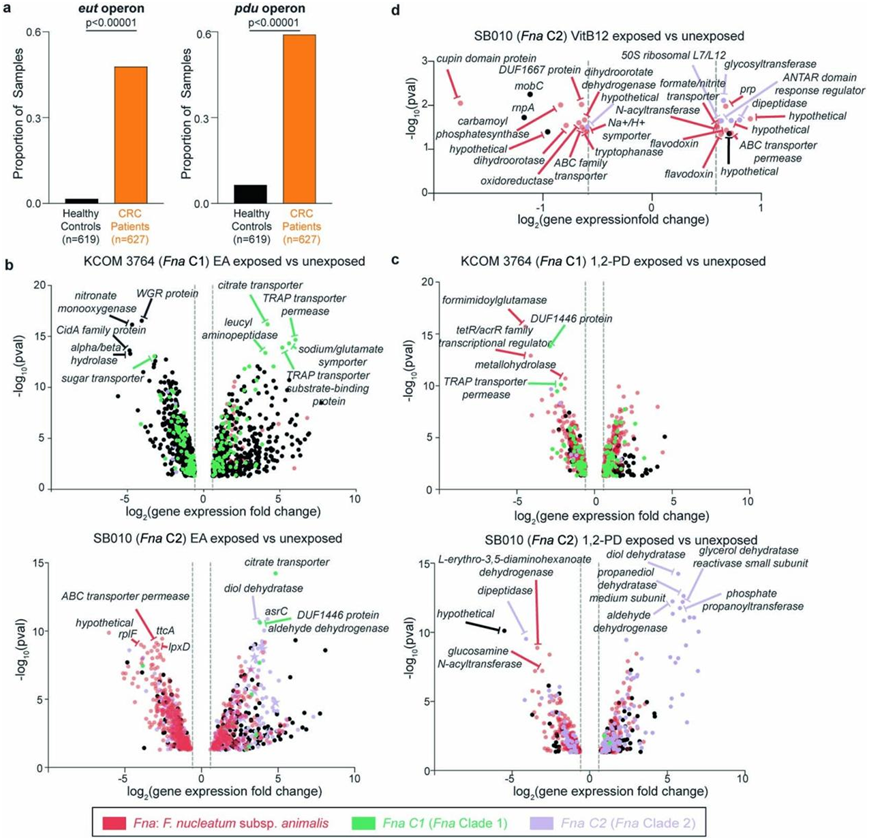
图S4. Fna分支对肠道代谢物的转录组反应。
(a) 条形图显示了来自 CRC 患者或健康对照的粪便宏基因组样本中检测到假定的eut和pdu操纵子的比例。 (b-d)上调和下调的基因分析。
04
Fna C2影响肠道肿瘤发生
由于富含Fna C2 的基因聚类主要与增强的代谢潜力相关(图S3e),在右旋糖酐硫酸钠诱导的结肠炎ApcMin+/- CRC小鼠模型中,他们试图确定Fna处理是否能够影响肠道肿瘤发生和体内代谢途径(图4a)。为了捕获更高比例的Fna进化枝特异性辅助基因(图S2d),他们对每个进化枝使用了三种代表性菌株的混合物。在单次口服Fna C1 混合物、Fna C2 混合物或载体对照后,到与Fna C1 和载体对照相比,Fna C2 处理的小鼠肠道腺瘤数量显著增加,特别是在大肠中(图4b)。Fna C1 处理小鼠和载体对照小鼠之间的腺瘤负荷没有显著差异。
他们对每个处理组的肠道组织进行了代谢组学分析。肠道代谢物的偏最小二乘判别分析表明,Fna C2 处理的小鼠形成了一个与其他处理组不同的聚类,表明代谢特征存在差异。然而,Fna C1 处理小鼠和载体对照小鼠的肠道代谢物具有更相似的代谢谱,聚集在一起(图4c)。对比分析显示,与Fna c1处理和对照小鼠相比,Fna c2处理小鼠的谷胱甘肽代谢和γ-谷氨酰氨基酸途径显著富集(图S7a-c)。具体来说,他们观察到γ-谷氨酰半胱氨酸(GSH)合成的前体水平显著增加,包括半胱氨酸和γ-谷氨酰半胱氨酸,还原型谷胱甘肽(GSH)水平下降,GSH降解产物5-氧脯氨酸水平显著升高(图S8a-c)。GSH 缺乏或氧化型 (GSSG) 与还原型 (GSH) 谷胱甘肽的比例升高会增加哺乳动物细胞对氧化应激、炎症和肿瘤进展的脆弱性。与对照组和Fna C1 处理组相比, Fna C2 处理组小鼠的 GSSG/GSH 比率显著增加,表明氧化应激增加(图S8d)。γ-谷氨酰转肽酶代谢GSH可以发挥促氧化作用。
在Fna c2处理的小鼠中,观察到其他氧化应激标志物(包括胱氨酸和半胱氨酸-谷胱甘肽二硫)水平显著升高,而能够清除活性氧的多胺(包括腐胺、亚精胺和精胺)水平显著降低(图S8b)。除了抗氧化应激作用外,多胺还能通过抑制巨噬细胞细胞因子的合成来抑制炎症。与炎症增加一致,与其他处理组相比,Fna c2处理小鼠中n -单甲基精氨酸和二甲基精氨酸水平显著升高(图S8b)。这两种代谢物都抑制抗炎剂一氧化氮的合成。此外,他们观察到促炎前列腺素和神经酰胺水平显著升高,包括前列腺素A2、n -棕榈酰鞘氨醇和n-棕榈酰鞘氨二嘌呤(图S8b)。神经酰胺还可由癌细胞代谢,以减少肿瘤细胞的凋亡和增殖。其他促进癌细胞增殖和转移的代谢物包括类二十烷酸,与Fna c1处理或对照小鼠相比,Fna c2处理小鼠的类二十烷酸也同样显著增加(图S7a-c,图S8e)。这包括通过COX2(也称为PTGS2)代谢花生四烯酸而增加6-酮前列腺素F1-α水平(图S8b)。值得注意的是,COX2是fn相关的人类结直肠肿瘤中表达上调最多的基因之一。
总的来说,这些研究结果表明Fna C2(而不是Fna C1)能够代谢影响肠道环境的促癌条件。
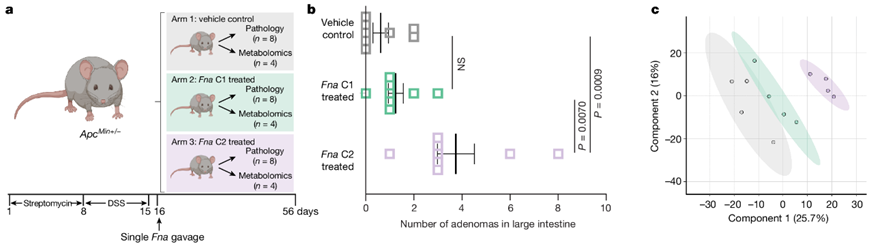
图4. Fna C2 对肠道肿瘤发生和代谢的影响。
(a) 小鼠模型建立示意图。(b) 显示处理组大肠腺瘤数量的图。(c) 检测到的肠道代谢物的偏最小二乘判别分析。
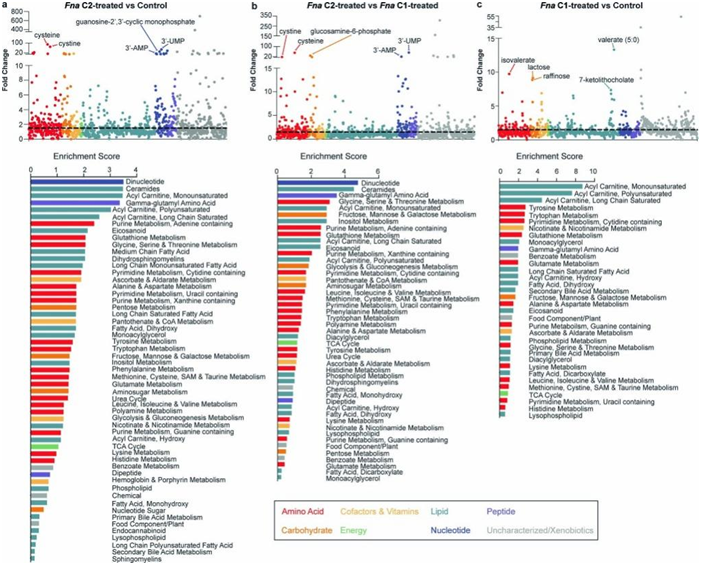
图S7. Fna处理小鼠的肠道代谢物变化。
(a-c)不同组中小鼠的代谢物变化分析。
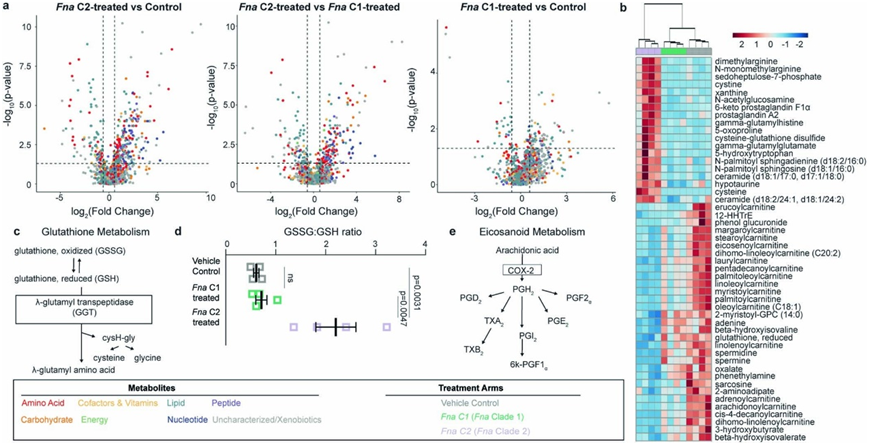
图S8. Fna处理小鼠的代谢物发生改变。
(a) 处理组之间代谢物水平变化分析。(b) 研究组中前 50 名代谢物的聚类热图。树状图根据代谢物谱的相似性对各个样本进行分组。(c) 谷胱甘肽代谢途径示意图。(d) 氧化型 (GSSG) 和还原型 (GSH) 谷胱甘肽水平之间的比率。 (e) 类二十烷酸代谢途径示意图。
05
人类 CRC 队列中Fna C2 富集
由于Fna C2 菌株在 CRC 生态位中显著富集(图2f),并且与Fna C1相比,在小鼠模型中增加了肠道肿瘤发生(图4b),因此他们接下来试图确定患病率以及在检测人体组织和粪便标本中这些Fna分支的丰度。
他们对 116 名初治 CRC 患者(CRC 队列 1)的切除肿瘤组织和其中 62 名患者的邻近正常组织进行了细菌 16S rRNA 测序。比较配对肿瘤和邻近正常组织之间不同梭杆菌属物种的相对丰度百分比 ,他们观察到与邻近正常组织相比, Fn是肿瘤组织中唯一显著富集的梭杆菌属物种(图5a)。然而,使用Fna进化枝特异性扩增子序列变体将Fn解析为更高的分类分辨率,包括Fna C1、Fna C2 和Fn的非Fna亚种,证明与配对的正常组织相比,只有Fna C2 在肿瘤中显著富集(图5a)。
在两个独立的患者队列(CRC队列1和CRC队列2)中,他们证明在CRC肿瘤组织内,与Fna C1相比,Fna C2显著富集(图5b)。
通过Fna C1 和Fna C2的随机效应模型对标准化均值差异进行荟萃分析表明,两个Fna分支均具有与 CRC 相关的显著合并效应大小(图5c)。然而, Fna C2的效应大小大于Fna C1。值得注意的是,在没有Fna C2 共现的情况下,Fna C1与 CRC 没有显著相关性(图5c)。Fna C2 在 CRC 患者的粪便中比Fna C1 更普遍和丰富(图5d)。
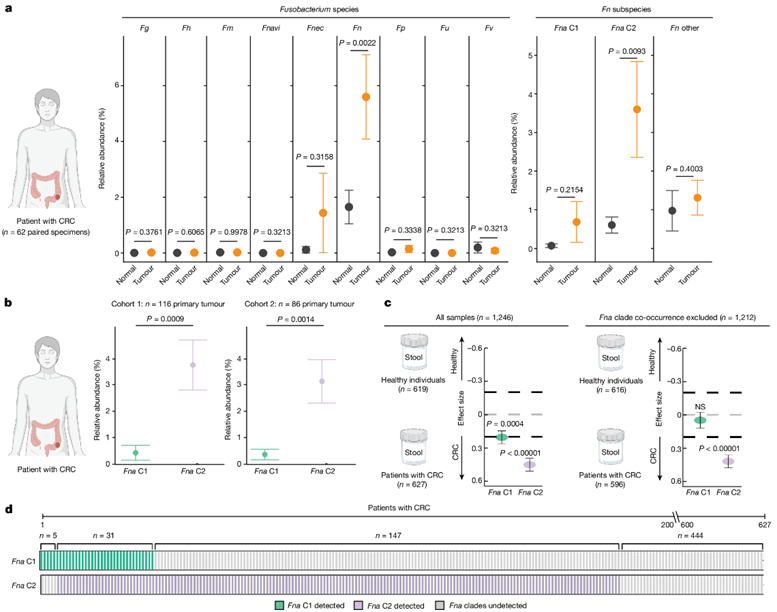
图5. 人体组织微生物组和粪便宏基因组样本中的Fn 。
(a) 显示Fn的相对丰度(左),以及Fn亚种和Fna进化枝(右)配对肿瘤(橙色)和正常邻近组织(黑色)的微生物 16S rRNA 基因测序。 (b) 图显示来自两个独立队列(队列 1 -本研究;队列 2-BioProject PRJNA362951)的患者原发性结直肠肿瘤组织中Fna C1(绿色)和Fna C2(淡紫色)的相对丰度。(c) CRC患者和健康个体粪便宏基因组数据中的Fna C1和Fna C2检测。(d) CRC 患者粪便宏基因组中存在Fna C1 和Fna C2。
+ + + + + + + + + + +
结 论
为了描述促进肿瘤定植的Fn遗传因素,本项研究生成了 135 个Fn菌株的基因组数据;来自非癌症个体的 80 种口腔菌株和从 51 名 CRC 患者的肿瘤中培养的 55 种独特癌症菌株。泛基因组分析确定了 483 个富含 CRC 的遗传因子。肿瘤分离菌株主要属于Fn亚种(Fna)。然而,基因组分析表明,Fna是单一亚种,但却由两个不同的进化枝(Fna C1 和Fna C2)组成。其中,只有Fna C2 在 CRC 肿瘤生态位中占主导地位,确定了 195 个Fna C2 相关遗传因素,这些因素与代谢潜力增加和胃肠道定植一致。Fna C2 处理的小鼠肠道腺瘤数量增加,代谢物发生改变。对 116 名 CRC 患者的人类肿瘤组织进行微生物组分析,结果显示Fna C2 富集。62 个配对样本的比较表明,与正常邻近组织相比,只有Fna C2 在肿瘤中富集。对 627 名 CRC 患者和 619 名健康个体粪便样本的宏基因组分析进一步支持了这一点。总的来说,本项研究结果确定了Fna进化枝分叉,表明Fna C2 特别驱动了人类 CRC 中的Fn富集,并揭示了Fna C2 对 CRC 生态位的病理适应的遗传基础。
+ + + + +



